Article Text

Abstract
Introduction Severe sialorrhoea is a common, distressing problem in children/adolescents with neurodisabilities, which has adverse health and social consequences. The SALIVA trial is designed to evaluate the efficacy and safety of a paediatric-specific oral solution of glycopyrronium along with its impact on quality-of-life (QoL), which has been lacking from previous trials of sialorrhoea treatments.
Methods and analysis A double-blind, placebo-controlled, randomised phase IV trial is ongoing in several centres across France. Eighty children aged 3–17 years with severe sialorrhoea (≥6 on the modified Teachers Drooling Scale) related to chronic neurological disorders in whom non-pharmacological standard of care has already been implemented or has failed, will be recruited. Patients will be randomised 1:1 to receive a 2 mg/5 mL solution of glycopyrronium bromide (Sialanar 320 µg/mL glycopyrronium) or placebo three times daily during a 3-month blinded period. After Day 84, participants will be invited into a 6-month, open-label study extension period, where they will all receive glycopyrronium. The primary endpoint of the double-blind period will be the change from baseline to Day 84 in the Drooling Impact Scale (DIS), a validated measure to assess sialorrhoea. A series of secondary efficacy endpoints involving change in total DIS, specific DIS items and response (DIS improvement ≥13.6 points) will be analysed in a prespecified hierarchy. QoL data will be collected from parents, caregivers and patients where possible using specific DIS questions and DISABKIDS questionnaires. Safety endpoints, including adverse events, will be assessed throughout the trial periods.
Ethics and dissemination In total, 87 children have been recruited and recruitment is now complete. Final results are expected by the end of 2023. Findings will be presented at conferences and published in peer-reviewed journals.
Trial registration number EudraCT 2020-005534-15.
- Therapeutics
Data availability statement
No data are available. Data sharing is not applicable as this manuscript describes a protocol. The trial is ongoing at the time of submission.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Severe sialorrhoea is a common and distressing condition in paediatric patients with cerebral palsy and other neurological diseases, which negatively impacts their health, social interactions and quality of life (QoL).
Previously, treatments with limited efficacy data and poor tolerability were used off-label, with no licensed therapies available, and no data on the impact of treatment on QoL outcomes.
In small, short-term clinical trials, glycopyrronium was shown to provide effective and well-tolerated improvement in chronic drooling and a novel paediatric-specific oral formulation has now been specifically approved for paediatric use in Europe.
WHAT THIS STUDY ADDS
The randomised double-blind, placebo-controlled SALIVA trial will provide important information on the efficacy of the EU-licensed oral liquid formulation of 320 µg/mL glycopyrronium using validated sensitive drooling scales and will also assess safety and the impact of treatment on QoL.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The results of this study will guide clinical decision-making for the treatment of severe drooling in children with neurodevelopmental disorders.
Introduction
Sialorrhoea or drooling is a common problem in children with neurodevelopmental disorders. Pathological drooling occurs in 40%–60% of children with cerebral palsy (CP)1–3 and is reported to be severe in 15% of cases.1 Dysfunction in oral-motor control appears to be the most important predisposing factor to drooling, and less frequently, over production of saliva (hypersalivation).4 5
Excessive drooling can result in skin irritation and infection, dehydration due to fluid loss, aspiration pneumonia and recurrent respiratory infections.3 5 Furthermore, drooling can adversely affect social interactions and self-esteem.6 7 As such, drooling has been shown to impact on health-related quality-of-life (QoL) in children with CP, influencing both physical health and psychosocial health QoL scores.7 Excessive drooling also places additional demands on caregivers, including frequent changes of clothing and bibs, and can damage equipment.8
Despite the considerable burden, the effective treatment of drooling remains a challenge. Only a relatively small number of clinical trials have been conducted and few licensed treatments are available for the paediatric population. Traditionally, conservative non-pharmacological rehabilitation is used as the first approach prior to anticholinergic drugs being considered.5 9 Botulinum toxin injection and surgical methods are alternatives when other strategies have failed.
Anticholinergics used in the treatment of drooling include atropine, benzhexol hydrochloride, benztropine, scopolamine patches and glycopyrronium.5 10 Due to non-selectivity, undesirable central and peripheral effects are frequently reported with anticholinergics, including sedation, irritability, headache, constipation, urinary retention and flushing.5 11 Factors such as the volume of liquid per dose,12 excipients and local tolerability issues also affect compliance and hence efficacy, safety and QoL. As a consequence of its quaternary charge, glycopyrronium has limited ability to penetrate the blood brain barrier compared with other anticholinergics resulting in fewer central adverse events (AEs).13 Limited trial data are available on the comparative efficacy and safety of anticholinergics; however, in the DRI trial and in a real-world study of children with developmental disabilities, glycopyrronium performed best in terms of reducing drooling with fewer AEs.10 14 The most common AEs with glycopyrronium are dry mouth (11%), constipation (20%), diarrhoea (18%), vomiting (18%), urinary retention (15%), flushing (11%) and nasal congestion (11%).15 QoL assessments have been lacking in the limited number of trials assessing therapeutic options.
Treatments for sialorrhoea had been mostly used off-label, but in 2016, the European Medicines Agency approved a novel paediatric-specific (in terms of concentration, excipients and licence) liquid formulation of 2 mg/5 mL glycopyrronium bromide (Sialanar 320 µg/mL glycopyrronium) to treat severe sialorrhoea in children (aged ≥3 years) and adolescents under a Paediatric Use Marketing Authorization.15
Here, we present the protocol of the SALIVA (Sialanar plus orAl rehabiLitation against placebo plus oral rehabilitation for chIldren and adolescents with seVere sialorrhoeA and neurodisabilities) trial, which is designed to evaluate the efficacy and safety of the 2 mg/5 mL oral glycopyrronium bromide formulation and also to assess its impact on QoL.
Methods
Trial design
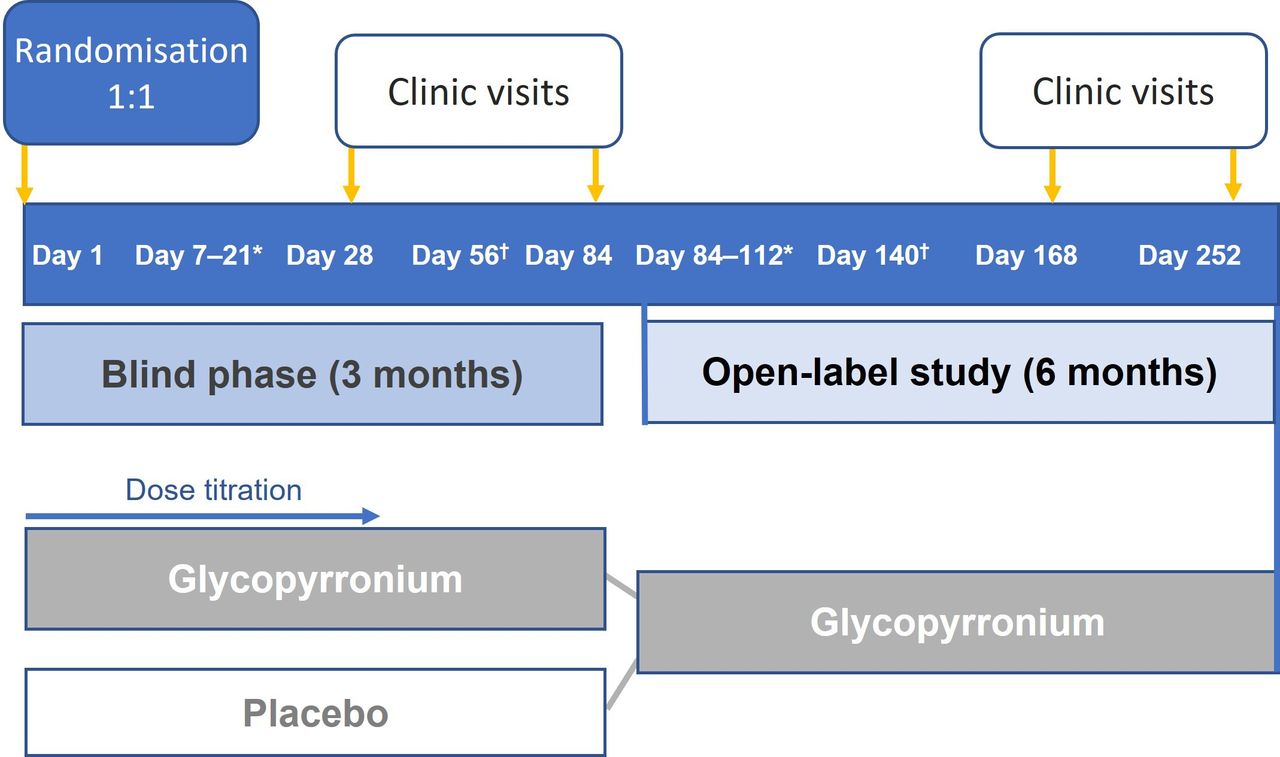
This is a double-blind, randomised, clinical phase IV trial comparing the oral glycopyrronium formulation (Sialanar) with placebo, in addition to continued non-pharmacological rehabilitation. Thirteen centres in France recruited participants and this recruitment is now complete. The trial consists of a 3-month main blinded period and a 6-month open-label extension (figure 1).

Trial design overview. *Telephone interviews once a week. †Telephone interview.
Trial population
Eligibility criteria are presented in table 1. Investigators considered new and existing patients for possible enrolment and obtained written consent from both parents (or the participant’s legally acceptable representative(s) where applicable) before recruitment. Children with chronic neurodisabilities (CP, Angelman syndrome, Rett’s syndrome, epilepsy, amyotrophic lateral sclerosis and intellectual disability) were eligible to enrol if they were aged between 3 and 17 years old and had severe sialorrhoea (defined as ≥6 on the modified Teachers Drooling Scale) and a Drooling Impact Scale (DIS) score ≥50. All participants had received ≥3 months of non-pharmacological rehabilitation, according to the standard of care outlined in French guidelines,16 and will continue to receive the same regimen during the trial. Children were not eligible if they received any anticholinergic therapy in the previous 4 weeks, botulinum injection within 6 months or surgery for drooling in the previous 12 months.
Eligibility criteria
Randomisation and intervention
Using an Interactive Web Response System, eligible participants were randomised 1:1 to receive either the oral glycopyrronium formulation or a matched placebo oral solution, three-times daily in a blinded manner for 3 months (figure 1). Placebo was selected as the comparator as, at the time of designing the trial, no licensed treatments for severe sialorrhoea were available in France. The dose of study drug will be titrated as needed over the first 4 weeks, consistent with the Summary of Product Characteristics (SmPC) of the active drug. Similarly, following the dose titration period, as per the SmPC, the child’s sialorrhoea will be monitored by the healthcare professional, in conjunction with the carer, to assess changes in efficacy and/or tolerability over time, and the dose adjusted accordingly. At enrolment, instructions were given to carers that treatment should be stopped and they should seek advice from the investigator in the event of constipation, urinary retention, pneumonia, allergic reaction, fever, very hot weather or changes in behaviour. After evaluating the event, the investigator will decide if treatment should remain stopped or a lower dose used. The parent/carer has been instructed to complete a notebook daily to record the dosage used and any AEs. Outpatient visits will take place at Day 28 and Day 84. Telephone interviews will occur every week during the titration period and at Day 56. All data are collected by the investigator and their study team at the study site. Pseudonymised data are entered by site personnel into a secure and validated electronic Case Report Form that is managed by the delegated contract research organisation with strict adherence to European Union General Data Protection Regulation. Identifiable, sensitive information is located securely at the study sites and is made available to delegated members of the study team to verify accuracy of data entry only.
After the Day 84 assessment, patients will be invited to continue into a 6-month open-label study extension (OLSE) where all patients will receive glycopyrronium. For those patients who previously received placebo, a period of titration will be performed. Clinic visits are scheduled for Day 168, with a final visit on Day 252.
Endpoints
The primary endpoint is the change in DIS score between baseline and Day 84. The DIS is a validated and reliable subjective measure that has been shown to be responsive to changes17 and has been used in other drooling studies.1 10 14 The French version, used in the SALIVA trial, is validated and has good test-retest reliability.18 The DIS is specifically designed to quantify the short- to medium-term treatment impact of saliva-control interventions. The questionnaire consists of 10 questions that are rated between 1 and 10 on a Likert scale covering efficacy and QoL assessments (figure 2). Items are completed by the investigator in an interview fashion with the same parent/carer (where possible) who has frequent and consistent contact with the patient. Scores are totalled to give an overall numerical rating of the severity and impact of drooling over the previous week. The lowest score is 10 and the maximum possible score is 100, with higher scores indicating greater severity and impact. The minimally clinically relevant difference was selected to be 13.6 points based on the findings of Reid et al.17 DIS items are related to the frequency and severity of drooling, the number of bib or clothing changes per day and information gained from parents and carers such as skin irritation, saliva smell, frequency of mouth wiping and the amount of saliva cleaning from household items (figure 2). Other items deal with embarrassment about dribbling and the impact of drooling on the child’s and family’s daily life.

{kind=link}
{kind=link}
Secondary efficacy endpoints are change in DIS between baseline and Day 28, the proportion of responders (DIS improvement ≥13.6 points) at Days 28 and 84, the proportion of good responders (DIS improvement ≥28 points based on Reid et al17) at Day 84, and changes in the number of used bibs or clothing over 7 days (DIS Item 3) at Days 28 and 84. Secondary QoL endpoints include change from baseline to Days 28 and 84 in DIS Item 9 (“To what extent did your child’s drooling affect his or her life?”) and in DIS Item 10 (“To what extent did your child’s dribbling affect you and your family’s life?”).
An additional QoL endpoint, change in DISABKIDS score from baseline to Day 84 and to Day 252, has been included in an attempt to assess whether an improvement in drooling can be measured in terms of overall QoL using a validated scale. DISABKIDS questionnaires are designed to assess health-related QoL in children and adolescents with chronic diseases19 rather than with neurodisabilities. The 37-item DISABKIDS Chronic Generic Measure for parents describes six dimensions (independence, physical limitation, emotion, social inclusion, social exclusion and treatment) with a 5-graded Likert scale transformed to numerical values (1–5), where higher values indicate better health-related QoL. A short 12-item version is designed for children aged ≥8 years, while the 6-item DISABKIDS Smiley Measure is designed for children aged 4–7 years or children who have not reached the level of reading ability necessary to complete the generic DISABKIDS questionnaire. Measuring QoL for children with drooling associated with neurological disabilities is challenging as a result of differences in their capabilities to complete an assessment due to their age and range of their abilities. Nevertheless, the measure has been included to establish whether it is sufficiently sensitive to detect an effect of drooling treatment in this patient population.
Regarding safety, collection of AEs started at the time of informed consent and will continue to be collected from the parent/carer and participant, where possible, at every visit (plus outside of visits in the daily notebook, as needed) until the final visit of the OLSE. Safety endpoints include AEs recorded from baseline to Day 84, including all AEs, all AEs except for dose-dependent expected AEs related to titration, AEs leading to discontinuation of study medication, all serious AEs, all treatment-related AEs and all treatment-related AEs except for dose-dependent expected AE related to titration.
Endpoints in the open-label extension phase include changes in DIS from baseline to Day 252 and from Day 84 to 252 in the glycopyrronium arm and between Day 84 to 252 for patients previously receiving placebo. QoL endpoints include change from baseline to Day 252 in DIS Items 9 and 10 and in the DISABKIDS questionnaires. All AEs from Days 84 to 252 will be recorded.
Statistical methods
A sample size of 23 subjects per group was calculated to be required to detect a minimal clinically significant difference of 13.6 points in the mean DIS score with 90% power, assuming a two-sided type 1 error rate of 5% and 13.6 as SD. Allowing for approximately 20%–30% loss to follow-up, enrolment of 60 children was estimated to be required to evaluate the primary endpoint. However, target enrolment was set at 80 children to compensate for terminations and to have an expected number of 60 children continue into the extension phase.
The primary endpoint will be evaluated in the full analysis set (intention to treat (ITT); which includes all randomised patients analysed according to the treatment they were randomised to receive) and in the modified ITT set (excludes all patients deemed ineligible after randomisation or who did not start study medication). Mean DIS score differences will be compared between arms through univariate analysis using Student t-tests considering the result of the equality of variances testing. Sensitivity analyses will include comparisons of the means of score differences (i) for patients with a DIS completed strictly by the same person at Day 0 and 84, (ii) when an unavailable DIS at Day 84 is replaced by the latest available DIS and (iii) in the per-protocol population (all patients who do not violate the terms of the protocol in a way that would affect the study outcome significantly, as determined by the study clinician blinded to study drug assignment).
For the primary criteria, a single statistical test will be done to control the alpha risk. Exploratory comparative analysis of DIS score change at Day 84 (±5 days) between the two treatment arms, adjusting for baseline DIS score, will be performed with analysis of covariance. Secondary efficacy endpoints will be explored. To address the issue of multiple testing and to limit the inflation of the alpha risk, a hierarchical test sequence is planned for these endpoints in the order: proportion of responders at Day 84, changes in DIS at Day 28, proportion of responders at Day 28, proportion of good responders at Day 84, changes in bib/clothes over 7 days at Day 84 and changes in bib/clothes over 7 days at Day 28. The secondary efficacy endpoints will be tested individually, in that order, if the statistical test on the primary endpoint is significant and until the first non-significant difference is found between the two treatment groups. All secondary endpoints will be described but once significance is lost, any additional analyses will be exploratory in nature and not confirmatory since no statistical test will be applied. No multiplicity adjustment will be made to the confidence intervals. The same analysis methods will be used for secondary endpoints as for the primary endpoint, with the χ2 test used for the responder analysis.
The analysis of QoL scores change over time between treatment arms will be performed as a repeated-measures analysis using all available timepoints. The results from mixed analysis of variance models will be used to assess mean difference and significance in DIS Item-9 score, DIS Item-10 score and total DISABKIDS scores, between treatment arms, considering each timepoint assessment.
All AEs, regardless of causality with study drug or seriousness, that occur from the first intake of study treatment until the end of study or until 3 days after the last intake of study treatment) will be considered in descriptive analyses.
In the OLSE, the endpoints will be described overall and according to the ex-treatment arm (ex-glycopyrronium arm or ex-placebo arm). AEs recorded from Day 0 to Day 252 will be analysed for all subjects who were assigned glycopyrronium for the entire period. Descriptive analyses will be performed and summary tabulations presented. No statistical test will be applied in the OLSE analysis.
Trial oversight
The sponsor (Proveca) participated in the design of the trial and is overseeing its conduct with a Steering Committee of Pierre Fayoux (Principal Investigator), Stéphane Auvin and Mickael Dinomais in conjunction with medical expert, Denis Pouchain. The trial is being conducted at the following sites (with lead clinicians shown in brackets): Jeanne de Flandre Hospital, Lille (Pierre Fayoux); Hôpital Robert Debré, Paris (Stéphane Auvin); CHU Angers-Les Capucins, Angers (Mickael Dinomais); Hôpitaux de Saint-Maurice, Paris (Aurélie Keslick); ESEAN Nantes (Guy Letellier); HFME l’Escale – CHU Lyon (Claire Mietton); MPR Dpt - CHU Grenoble (Véronique Bourg); CHU Poincaré -APHP Garches (Delphine Verollet); CAMPS-CHU Rouen (Stéphane Rondeau); CHU Strasbourg (Vincent Laugel); CHU La Timone APH—Marseille (Béatrice Desnous); CHU Saint-Etienne (Vincent Gautheron) and CMCR Les Massues Lyon (Fabienne Roumenoff).
Trial status
The trial is ongoing. Recruitment started in May 2021 and is now complete, with 87 children enrolled. There have been no major protocol amendments except those related to the COVID-19 pandemic, namely, extension of the inclusion period and the requirement for the negative COVID-19 test at enrolment. Primary results of the double-blind period are expected in mid-2023 and final results are expected by the end of 2023.
Conclusion
Severe sialorrhoea places a considerable burden on children with neurodisabilities and on their families. Few licensed treatments are available and clinical trial data are relatively scarce. The SALIVA trial provides an opportunity to strengthen knowledge on the effects of a licensed glycopyrronium formulation (Sialanar) in terms of efficacy, using validated sensitive scales, with regard to tolerability during titration and with long-term use and also by formally evaluating QoL using specific tools.
Data availability statement
No data are available. Data sharing is not applicable as this manuscript describes a protocol. The trial is ongoing at the time of submission.
Ethics statements
Patient consent for publication
Ethics approval
The protocol has been approved by an independent ethics committee and the French Agence Nationale de Sécurité du Médicament. Participants gave informed consent to participate in the study before taking part.
Footnotes
Twitter @proveca
Contributors PF (Principal Investigator), SA, MD and DP are part of the Steering Committee; they participated in trial design and supervised drafting of the manuscript. HS, NP and FV participated in trial design and manuscript preparation. NT coordinates the trial, data collection and analysis. All authors approved the final draft of the manuscript.
Funding The trial is funded by Proveca Ltd—no award/grant number. In the drafting of the protocol manuscript, editorial assistance was provided by Emma Marshman and funded by Proveca Ltd.
Competing interests PF receives fees from Merz Pharma for providing training sessions. SA has served as consultant or given lectures for Angelini, Biocodex, Eisai, Encoded, Grintherapeutics, Jazz Pharmaceuticals, Neuraxpharm, Orion, Nutricia, Proveca, UCB Pharma, Vitaflo, Xenon and Zogenix. SA has been an investigator for clinical trials for Eisai, Marinus, Proveca, Takeda, UCB Pharma and Zogenix. MD and DP report no competing interests. HS, NP and FV are employees of Proveca Ltd. NT is an employee of the contract research organisation, Kappa Santé.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.