Article Text

Abstract
Introduction Haemodynamically significant patent ductus arteriosus (hsPDA) is a common cause of mortality and morbidity in preterm infants. Existing medical therapies with ibuprofen or indomethacin have multiple adverse effects. Hence, an alternative drug like paracetamol given through oral route with less side effects need to be tested in an appropriate study design with least risk of bias to arrive at a conclusion.
Methods and analysis Multisite, randomised, active-controlled, non-inferiority design. The primary objective is to study the efficacy of oral paracetamol for closure of hsPDA in comparison to oral ibuprofen in preterm neonates of <32 weeks’ gestation. Randomisation web-based and allocation concealment would be done; the treating team, investigators, outcome assessors and laboratory personnel would be blinded from the intervention. Echocardiography images would be coded for independent review. Closure of PDA by the end of last dose of study drug or earlier would be the study endpoint. A sample size of 196 neonates would be enrolled with a non-inferiority margin of 15%. Both intention-to-treat and per-protocol analysis will be done to assess the effect of contamination and protocol violations in the primary outcome.
Ethics and dissemination The trial would follow international code of ethics for clinical trial. The trial protocol was approved by the Institute Ethics Committee of all three centres. All serious adverse events would be reported in detail to the Institute Ethics Committee. A written informed consent would be obtained from one of the parents. No plan has been made for dissemination.
Trial registration number CTRI/2014/08/004805.
- cardiology
- circulatory
- imaging
- intensive care
- neonatology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
What is already known on this topic?
Ibuprofen and indomethacin are the current standard drugs for closure of a haemodynamically significant patent ductus arteriosus (PDA) apart from surgical ligation.
These drugs have many adverse effects involving the gut, kidneys and the pulmonary vasculature.
Few case reports and two clinical trials have reported about use of oral paracetamol for closure of PDA.
What this study hopes to add?
We intend to compare oral paracetamol with oral ibuprofen to demonstrate that paracetamol is not inferior in efficacy and is safer in comparison to ibuprofen.
Being one of the largest multisite, non-inferiority trial where the investigators, treating team, laboratory personnel and the outcome assessors would be blinded to the intervention is a major strength of this study.
Study background and rationale
Haemodynamically significant patent ductus arteriosus (hsPDA) is a common cause of morbidity and mortality in preterm neonates.1 2 Treatment options for the closure of hsPDA include pharmacological therapy and surgical ligation. Indomethacin and ibuprofen, both inhibit the conversion of arachidonic acid to prostaglandins, are the two most commonly used drugs for closure of PDA.3 4 Ibuprofen has been reported to successfully close hsPDA in 70%–85% of cases.5–8 However, several serious adverse effects have been reported with both indomethacin and ibuprofen, which include intense peripheral vasoconstriction, gastrointestinal bleeding and perforation, decreased platelet aggregation, hyperbilirubinaemia and renal failure.9 10 Paracetamol (also known as acetaminophen), unlike ibuprofen, acts on prostaglandin synthase at the peroxidase region of the enzyme.11 Paracetamol-mediated inhibition of the peroxidase region enzyme was reported to be further facilitated by decreased local concentration of hydroperoxides.12 The role of paracetamol as an alternative treatment for closure of hsPDA has gained attention in recent years because of its superior safety profile in comparison to the cyclooxygenase inhibitors.13–15 Five case series together consisting of 39 neonates who received paracetamol for significant PDA (where indomethacin and ibuprofen were contraindicated) had reported a closure rate of 84%–100%.13 14 16 17 A clinical trial comparing oral paracetamol with oral ibuprofen in 80 preterm neonates concluded that oral paracetamol was not superior to oral ibuprofen,18 whereas another trial with 160 preterm neonates reported that oral paracetamol was not inferior to oral ibuprofen in closure of PDA.19 A Cochrane database systematic review of two low-quality unmasked studies that enrolled 250 infants (above two studies) showed no significant difference between treatment with oral paracetamol versus oral ibuprofen for failure of ductal closure after the first course of drug administration (typical relative risk (RR) 0.90, 95% CI 0.67 to 1.22).20 There were also no significant differences between the paracetamol and the ibuprofen groups in the secondary outcomes except for ‘duration for need of supplemental oxygen’ and for hyperbilirubinaemia, both in favour of paracetamol. However, both the studies included were graded down by the review group as low in quality using the Grading of Recommendations, Assessment, Development and Evaluation(GRADE) process. Another recent systematic review of 16 studies (2 randomised controlled trials and 14 uncontrolled studies) concluded that the efficacy and safety of paracetamol appear to be comparable with those of ibuprofen. However, the authors cautioned about the non-optimal quality of the studies analysed and the limited number of neonates treated with paracetamol so far and advised for additional well-designed studies to support the use of paracetamol for PDA in current clinical practice.21 A recent report has reinforced the long-term neurodevelopmental safety of paracetamol in comparison to ibuprofen in 80 preterm neonates.22 Considering the equivocal reports published until now and the promise offered by paracetamol as a safer alternative, this randomised, active controlled, masked, non-inferiority trial was planned.
Research question
Is oral paracetamol administered in a dose of 15 mg per kg per dose every 6 hours for 3 consecutive days associated with a rate of closure of PDA not inferior by a non-inferiority margin (D) of 15% in comparison to oral ibuprofen in a dose of 10 mg per kg per dose on day 1 and 5 mg per kg per dose at 24 and 48 hours from the first dose in preterm neonates (<32 weeks) with evidence of haemodynamically significant (HS) PDA?
Description of the study hypothesis
Null hypothesis (H0): T – S ≤ D
Alternate hypothesis (one-sided) (HA): T – S > D
T — rate of closure of PDA in the test group that would receive oral paracetamol
S — rate of closure of PDA in the standard, active control group that would receive oral ibuprofen
D — non-inferiority margin set at minus 15%
Objectives
Primary objective
The primary objective is to study the efficacy of oral paracetamol for closure of hsPDA in comparison to oral ibuprofen in preterm neonates of <32 weeks’ gestation with evidence of hsPDA.
Secondary objective
The secondary objective is to compare the following between oral paracetamol and oral ibuprofen groups:
Time to closure of PDA
Proportion of neonates where PDA closed following a single course
Proportion of neonates who required surgical ligation for closure of PDA
All-cause mortality before discharge from hospital
Proportion of neonates where the PDA reopened
Incidence of echocardiography-proven pulmonary artery hypertension
Duration of mechanical ventilation (in days)
Duration of any respiratory support (in days)
Duration of need for supplemental oxygen (in days)
Incidences of azotaemia, oliguria, hepatitis with deranged liver transaminases, deranged coagulogram, intraventricular haemorrhage (any grade of severity), severe intraventricular haemorrhage (grade 3 and intraparenchymal extension), periventricular leucomalacia, necrotising enterocolitis (all stages), necrotising enterocolitis (definite and advanced stage as per modified Bell’s staging), feed intolerance, bronchopulmonary dysplasia and retinopathy of prematurity.
Outcome measures
Primary outcome measure
The primary outcome measure is closure of PDA by the end of the last dose of the study drug or earlier, irrespective of the course of the drug.
Secondary outcome measures
Closure of PDA following a single course of study drug
Closure of PDA following surgical ligation
Death (due to any cause) before discharge from the hospital
Reopening of PDA following initial closure
Echo-proven pulmonary artery hypertension
Azotaemia, oliguria, hepatitis with deranged liver transaminases, deranged coagulogram, intraventricular haemorrhage (any grade of severity), severe intraventricular haemorrhage (grade 3 and intraparenchymal extension), periventricular leucomalacia, necrotising enterocolitis (all stages), necrotising enterocolitis (definite and advanced stage as per modified Bell’s staging), feed intolerance, bronchopulmonary dysplasia and retinopathy of prematurity.
Methodology
Study design
This is a randomised, two parallel arm, active-controlled, blinded, non-inferiority trial.
Study place
This study will be conducted at three centres namely - Division of Neonatology,Department of Pediatrics, Postgraduate Institute of Medical Education and Research,Chandigarh, India; Department of Neonatology, Fernandez Hospital, Bogulakunta, Hyderabad, India and Department of Neonatology, Institute of Child Health, Egmore, Chennai, India.
Eligibility criteria
Inborn preterm neonates <32 weeks’ gestation
Presence of a haemodynamically significant PDA*
*hsPDA is defined if any one of the below-mentioned clinical/biochemical sign is present in the presence of a PDA with a transductal diameter of ≥1.6 mm (or) in the presence of any one of the below-mentioned echocardiographic sign suggestive of haemodynamic significance even in the absence of any of the below-mentioned clinical/biochemical sign. A screening echo would be done in all asymptomatic neonates to detect hsPDA and this would be timed between 48–72 hours of age in infants 29–31 weeks and in the first 48 hours for that ≤28 weeks’ gestation.
Group A*: signs of significant left→right shunt: hyperdynamic pulsatile precordium, bounding peripheral pulses and wide pulse pressure (>25 mm Hg)
Group B: signs of systemic underperfusion: poor peripheral pulse volume, prolonged capillary refill time, decreased urine output, deranged renal function test, metabolic acidosis and hypotension
Group C: signs of pulmonary overperfusion: abnormal weight gain, increase in liver size, new onset or increase in ventilatory requirements that primarily involve Positive End Expiratory Pressure (PEEP) Peak Inspiratory Pressure (PIP) and Fraction of Inspired Oxygen (FiO2), respiratory acidosis, pulmonary crepitations and haemorrhagic pulmonary oedema
*In the presence of a clinical sign that falls under Group A, a second trained neonatologist would be asked to confirm the clinical sign and the sign would be present only if both examiners concur; wide pulse pressure must be recorded on two consecutive blood pressure measurements.
Echocardiographic features indicative of hsPDA:
A transductal diameter of ≥1.5 mm plus one of the following:
Evidence of left atrial enlargement (Left atrium: Aortic rootroot diameter ratio ≥1.4)
Ductal velocity <2 m/s
Antegrade main pulmonary artery (MPA) diastolic flow >20 cm/s
E wave: A wave ratio >1
Isovolemic relaxation time (IVRT) ≤45 ms
Absent or reversed diastolic blood flow pattern in descending thoracic aorta.
Exclusion criteria
Antenatally or postnatally suspected or diagnosed structural heart disease
Presence of major congenital malformations
Contraindication for enteral feeding
Contraindication for administration of any one of the study drugs such as blood urea >60 mg/dL, serum creatinine level >1.6 mg/ dL, platelet count <60 x 109/L, clinical bleeding from any site, deranged coagulogram, clinical or radiological evidence of necrotising enterocolitis, intraventricular haemorrhage of moderate to severe grade severity (grade III with or without intraparenchymal extension) or progression of intraventricular haemorrhage demonstrated in an earlier ultrasound, and hyperbilirubinaemia within 2 mg/dL from the exchange transfusion cut-off value
Refusal of consent.
Enrolment process
Consent
One of the parents would be approached for consent to allow their newborn infant to participate in this clinical trial (figure 1). The parents will be provided a detailed parent information sheet and will also receive a verbal explanation about the study. Neonates will be enrolled only after obtaining written informed consent from one of the parents. Parents would be allowed to withdraw their neonate from the study at any stage.

Study flow as per the Consolidated Standards of Reporting Trials (CONSORT) recommendations.
Intervention and comparison groups
Intervention
Paracetamol oral suspension (Calpol, GlaxoSmithKline Asia) would be administered through an orogastric tube in a dose of 15 mg/kg/dose at six hourly intervals for three consecutive days. The drug would be filled in 5 mL plastic syringes and would be gently pushed through the orogastric tube followed by a flush of 1 mL of sterile water for injection.
Active control
Ibuprofen oral suspension (Ibugesic, Cipla India) would be administered through orogastric tubes in a dose of 10 mg/kg/dose followed by 5 mg/kg/dose after 24 and 48 hours from the first dose. A similar drug administration technique would be followed as stated above.
Allocation process
Web-based random allocation would be done within each of the three strata (<28 weeks, 28–29 and 30–31 weeks) (http://www.randomization.com). A block randomisation would be done with blocks of variable sizes within each stratum. Separate personnel who are not involved in any aspect of the trial would do the random allocation.
Allocation concealment and blinding
The drugs would be prepared and dispensed by the clinical pharmacy department of the institute. The drugs would be dispensed in 5 mL volumes in separate phials as per the group of allocations. Allocation concealment would be ensured by serially numbering the phials with a randomly generated code number corresponding to the sequence of allocation. The drugs would be prepared to have a similar colour, flavour and viscosity to prevent identification of the study drug and would be dispensed on opaque plastic phials. The blinding process would be tested by administering the dispensed drugs to adult volunteers to assess whether they can differentiate the drugs based on the colour, flavour, taste and viscosity of the preparations. To avoid recognising the study drug due to differences in their respective dosage and frequency schedules, the concentration of the drugs would be modified in such a way that administering equal volume of the drug at a time point would ensure appropriate dosing of that drug for that time point. The final concentration that would be achieved would be 1 mL=15 mg for paracetamol and 1 mL=10 mg for ibuprofen. Moreover, as once daily schedule would be followed for ibuprofen, a similar resembling placebo (inert agent) would be used to complete a sham six hourly dosing schedule in the case of the ibuprofen group. This process would be ensured by having a set of 24 phials (12 phials for the first course and 12 more for the second course) for each enrolled neonate, with the phials clearly marked with the code number, sequence of enrolment and the day of therapy on the phial label. For the second course, phials marked days 4, 5 and 6 would be used. No drug from an already opened phial would be reused for the next day therapy. This whole process would keep the treating team, investigators, outcome assessors and the laboratory personnel blinded from the nature of the intervention. Moreover, the outcome assessors would be further blinded by coding the echo images stored in the system for assessment of ductal closure.
Determination of ductus arteriosus status by echocardiography
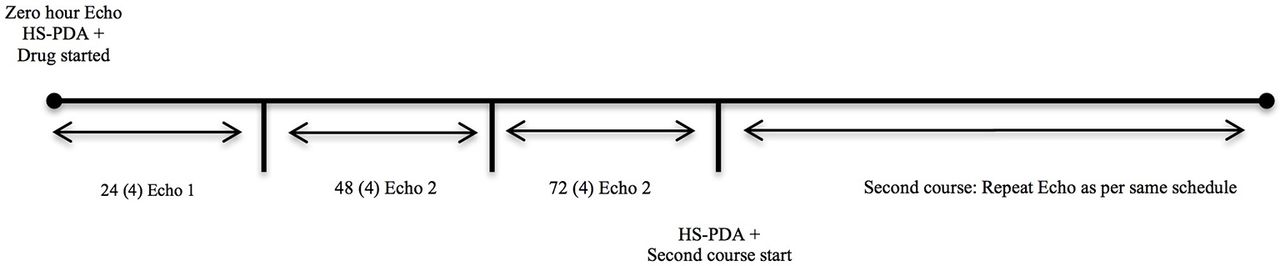
Daily echocardiographic assessments would be done until completion of the course or until the closure of the PDA, whichever is earlier (figures 2 and 3). A PDA would be considered as closed if there is no demonstrable open ductus and the colour Doppler demonstrates no flow across the ductus arteriosus region. After 24 hours from the completion or earlier in case clinical signs appear, a repeat echocardiogram would be done to assess for reopening of PDA. The echocardiogram images and clips would be code-numbered and archived for review VS or SSS. SD will maintain the key to the code numbers.

Operational flow and timeline of the study. hsPDA, haemodynamically significant patent ductus arteriosus.
{kind=link}
{kind=link}
{kind=link}
Timeline of assessments by bedside echocardiography. hsPDA, haemodynamically significant patent ductus arteriosus.
Technique of transthoracic echocardiography
The echocardiographic assessments will be done using a SonoSite MicroMaxx Portable Ultrasound Machine (Fujifilm SonoSite, Bothell, Washington, USA) in site 1, Philips CX50 machine (Philips Healthcare, Boston, Massachusetts, USA) in site 2 and Esaote MyLab Five (Esaote, Genova, Italy) in site 3. A cardiac probe of 8–12 MHz frequency will be used for the study. Before the procedure, the probe head will be cleaned and cold-disinfected with 2% glutaraldehyde solution. Three standard views would be optimised for visualisation of the ductus arteriosus: subxiphoid, high parasternal (ductal) and aortic arch (suprasternal) views. Presence of ductus in two dimensions (2D) would always be cross-checked using a colour Doppler superimposition. Transductal diameter would be measured preferably in 2D in a high parasternal cut. To maintain uniformity, the diameter measurement will be done at the narrowest point by 2D imaging, which is usually at the pulmonary end of the ductus arteriosus, where it constricts first. Colour Doppler mapping would be used to visualise the direction of shunt blood flow. Ductal velocity would be assessed using pulsed wave Doppler (PWD) positioned at the pulmonary end of the ductus. Similarly, PWD would be used to assess the antegrade flow in diastole in the MPA. M-mode would be used to measure the left atrial:aortic root diameter ratio. PWD with range gate would be placed transmitrally for the E:A ratio and between the mitral and aortic valves for IVRT. Left ventricular output (LVO), right ventricular output (RVO) and superior vena cava (SVC) flow would be measured using the following formula: [VTI (cm) × π × (D/2)2× heart rate]/birth weight (kg). Velocity time integral (VTI) will be calculated based on PWD across the left ventricle outflow tract for LVO, right ventricular outflow tract for RVO and SVC for SVC flow. The descending aorta would be visualised in the suprasternal view and using continuous wave Doppler the flow direction in diastole would be calculated for aortic run-off.
Training of the principal investigator
The principal investigator (PI) in each site would be formally evaluated over a period of 1 month preceded by a hands-on training session. During the training session, the PI would be trained by one of the coinvestigators who have experience in performing neonatal functional echocardiographic assessments (VS and SSS). During the evaluation period, the PI would record 50 images and video clips from 50 different neonates with 20% of them being normal neonates. VS or SSS would review these images for correctness of image acquisition and diagnosis. The PI would be considered trained to perform independent echocardiography for the research purpose when he gets >90% of the images correct. At the end of evaluation, 10 out of 50 images would be repeated by coinvestigators VS or SSS to check for interobserver variability.
Sample size estimation and statistical analysis
The observed rate of PDA closure by oral ibuprofen was 85%.23 Assuming an equal rate of closure for oral paracetamol with a non-inferiority margin of 15%, one sided alpha error of 5% and power of 90%, 196 neonates would be required in this trial. The primary outcome (binary) of rate of closure of PDA will be compared between the study groups by calculating the RR and risk difference and would be expressed as 95% CI. Baseline variables would be compared between the study groups using χ2 test for categorical variables and Student’s t-test or an appropriate non-parametric test for numerical variables. Time to closure of PDA would be compared by a Kaplan-Meier curve and significance will be tested by log-rank test. Both intention-to-treat and per-protocol analysis will be done to assess the effect of contamination and protocol violations on the primary outcome. Apart from hypothesis testing for non-inferiority using the non-inferiority margin assumed, the 95% CI of the probability of closure of PDA would be marked to establish or reject non-inferiority of paracetamol in comparison to oral ibuprofen. A ‘p’ value of 0.05 would be considered significant. IBM SPSS V.20 will be used for data entry and statistical analysis.
Ethics and dissemination
The trial would follow international code of ethics for clinical trial and good clinical practices. The trial protocol has been approved by the Institute Ethics Committee of all the three centres. All serious adverse events would be reported in detail to the Institute Ethics Committee. A written informed consent would be obtained from one of the parents. No plan has been made for dissemination.
References
Footnotes
Contributors AsK assisted in designing and planning the study and wrote the first draft of the protocol. VS conceived the idea, designed the trial and critically edited and approved the manuscript. TPO assisted in planning the study and critically edited and approved the manuscript. SM assisted in designing and planning the study and critically edited and approved the manuscript. ArK assisted in planning the study and wrote the first draft of the protocol. MS assisted in designing and planning the study and critically edited and approved the manuscript. SSS assisted in planning the study and critically edited and approved the manuscript. SD assisted in designing and planning the study and critically edited and approved the manuscript.
Funding This study and the manuscript did not attract funding from any source.
Competing interests None declared.
Patient consent This is a study protocol and hence consent form is not applicable at this stage.
Ethics approval Institute Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional unpublished data are available in this case as this is a protocol. Data that are being collected can be accessed by one of the site investigators (VS, SM and MS), and they should be preferably contacted through the corresponding author to obtain the data.