Article Text

Abstract
Background Congenital anomalies (CAs) are a common cause of infant death and disability. We linked children from a large birth cohort to a routine primary care database to detect CA diagnoses from birth to age 5 years. There could be evidence of underreporting by CA registries as they estimate that only 2% of CA registrations occur after age 1 year.
Methods CA cases were identified by linking children from a prospective birth cohort to primary care records. CAs were classified according to the European Surveillance of CA guidelines. We calculated rates of CAs by using a bodily system group for children aged 0 to <5 years, together with risk ratios (RRs) with 95% CIs for maternal risk factors.
Results Routinely collected primary care data increased the ascertainment of children with CAs from 432.9 per 10 000 live births under 1 year to 620.6 per 10 000 live births under 5 years. Consanguinity was a risk factor for Pakistani mothers (multivariable RR 1.87, 95% CI 1.46 to 2.83), and maternal age >34 years was a risk factor for mothers of other ethnicities (multivariable RR 2.19, 95% CI 1.36 to 3.54). Education was associated with a lower risk (multivariable RR 0.78, 95% CI 0.62 to 0.98).
Conclusion 98% of UK CA registrations relate to diagnoses made in the first year of life. Our data suggest that this leads to incomplete case ascertainment with a further 30% identified after age 1 year in our study. Risk factors for CAs identified up to age 1 year persist up to 5 years. National registries should consider using routine data linkage to provide more complete case ascertainment after infancy.
- comm child health
- congenital abnorm
- data collection
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
What is already known
Interrogating primary care data for Read medical codes is a valid and useful source for ascertaining disease prevalence in research studies.
Primary care data linked to prospective birth cohort studies allow for detailed, longitudinal analysis.
What this study adds
Continuing case attainment to age 5 years increased the prevalence of congenital anomalies (CAs) in our population, from 432.9 per 10 000 live births at age 1 year to 620.6 at age 5 years.
Without accurate case ascertainment, it is likely that both general and specialist services for CAs will be underresourced.
Introduction
Around 93% of children with a congenital anomaly (CA) survive infancy and will require varying levels of support from health services.1 Most registrations reported by CA registers in the UK occur very early in life, with less than 2% of registrations after 1 year of age.2 Out of the 36 CA registries in Europe, seven reported that more than 2% of cases were diagnosed after age 1 year. These later registrations amounted to between 5% and 10% of total CA registrations.3 Comparisons between registries is made more difficult by inconsistencies in the definitions and data variables between different registers and by an absence of follow-up data.4–8
This study uses detailed sociodemographic and clinical information from the Born in Bradford (BiB) prospective birth cohort. The incidence of CA in Bradford is high, previously reported at 306 per 10 000 live births, compared with a national average of 227 per 10 000 live births.2 9 10 The Bradford rate, however, is based on case ascertainment up to the child’s first birthday, as are 98% of cases used to calculate the national rate. We suspect this may lead to underascertainment of CA and a disparity in both need and demand for children’s healthcare. For example, delayed diagnoses have been reported in 10% of congenital heart defects,11 one of the most frequently diagnosed subgroups of CA nationally.2 10 12 Interrogation of diagnoses recorded in the General Practice Research Database (GPRD) also revealed the prevalence of heart defects to be nearly 50% higher than in European Registers of Congenital Anomalies (EUROCAT) projections, with a similar increase in prevalence in other system-specific subgroups also recorded.13
Higher overall CA prevalence rates have been recorded from primary care data sources in comparison with national databases, suggesting the utility of primary care data to serve as a more complete source of background prevalence.12 13 Several other studies report the potential of routine primary care databases for CA case ascertainment and diagnoses after age 1 year.5 12–17 These databases generally identify more cases than national registers and also have the advantage (over national registers) of including information on potential parental and child risk factors for CA and clinical information on the child across their life course, both of which can be used to improve understanding of the causes and consequences of CA.5 12–17 However, primary care databases may lack information on all diagnoses and will not have systematically collected data on potential risk factors using the same methods for all children.
Objective
Our aims were to compare case ascertainment of CA from birth to under 5 years between national CA rates and a pregnancy/birth cohort linked to primary care data. Our a priori hypotheses are that more cases will be ascertained from the primary care-linked birth cohort than national rates, and the detailed data in the birth cohort will allow us to determine whether magnitudes of association for risk factors persisted. We compare CA detection between the primary care database and the clinically diagnosed CA from medical records to determine the accuracy of the primary care diagnoses information.
Design
Setting
We used data from BiB, an ongoing prospective birth cohort study, which recruited 12 450 pregnant women between 2007 and 2011. The BiB methods are reported in detail elsewhere.18
Case ascertainment and coding methods
BiB recruits gave their consent to access electronic primary care records held on SystmOne,19 the patient contact single-source system that currently has complete coverage in Bradford and is linked to BiB baseline questionnaire data. This linkage provides a unique data set comprising detailed social, environmental and clinical data on the mothers and children in the study. Primary care data were extracted for each child when there was an exact match for the National Health Service (NHS) number, surname, date of birth and gender between SystmOne19 and BiB. Of 13 857 recruits, 97% were matched to primary care data, forming the study population. In all, there were 74 386 person-years of data, and the average time over which data were recorded in the primary care record was 5.5 years, with a maximum of 7.6 years. Not all children had reached age 7 years, so we censored our follow-up of these cases to under age 5 years. SystmOne19 assigns any diagnosis a CTV3 Read medical code. We mapped Read codes to the International Classification of Diseases, 10th Edition (ICD-10) codes to allow anomaly classification, with assignment to an anomaly group based on the system affected and syndrome (where applicable). We followed the EUROCAT guidelines,20 using the British Isles National Organisation of CA Registers (BINOCAR) methodology.2 A codebook of both major and minor CAs was mapped to Read codes extracted from the primary care database using cross mapping.21 Classifications were reviewed by a clinical geneticist (ES).
Sensitivity analyses
We validated the original mapping of CTV3 Read codes to ICD-10 by repeating the mapping process with different members of the research team. We recorded all cases up to the date the child left the primary care practice (ie, had died or moved away), the date the practice stopped recording primary care appointment data or the date of the child’s last appointment at time of data extraction (July 2016). A child was classified as having a CA if one or more Read codes for CA were recorded in the child’s primary care record at any time during which the child was registered at the practice. From combining CAs reported in the previous BiB data set10 (phase 1 ‘notifications’), which were children identified by a standard hospital notification system ages 0 to <1 year and confirmed by a clinical review, and CA identified in primary care data (phase 2 ‘data linkage’) ages 0 to <5 years, we found 296 CAs reported by both phase 1 and phase 2 methodologies. We were able to validate whether phase 1 and phase 2 diagnoses matched, an important step in determining whether clinical diagnoses made in hospital matched Read code entry into primary care databases (figure 1). We reached 83% agreement. We then calculated the prevalence of CA overall and for bodily system-specific subgroups for children diagnosed between ages 0 and <5 years. We also calculated prevalence up to age 1 year for comparison with EUROCAT registries. We also found 127 CAs (17%), which did not match between phase 1 and phase 2 methodologies. On further inspection, these cases had ICD-10 codes outside of the 27th June CA chapter as recommended by EUROCAT. The clinicians responsible for the phase 1 study explained this was due to some conditions being so rare they could not find an appropriate code within the recommended CA ICD-10 chapter. Other reasons included the death or moving primary care practice more than one time, causing potential errors in their diagnoses records.
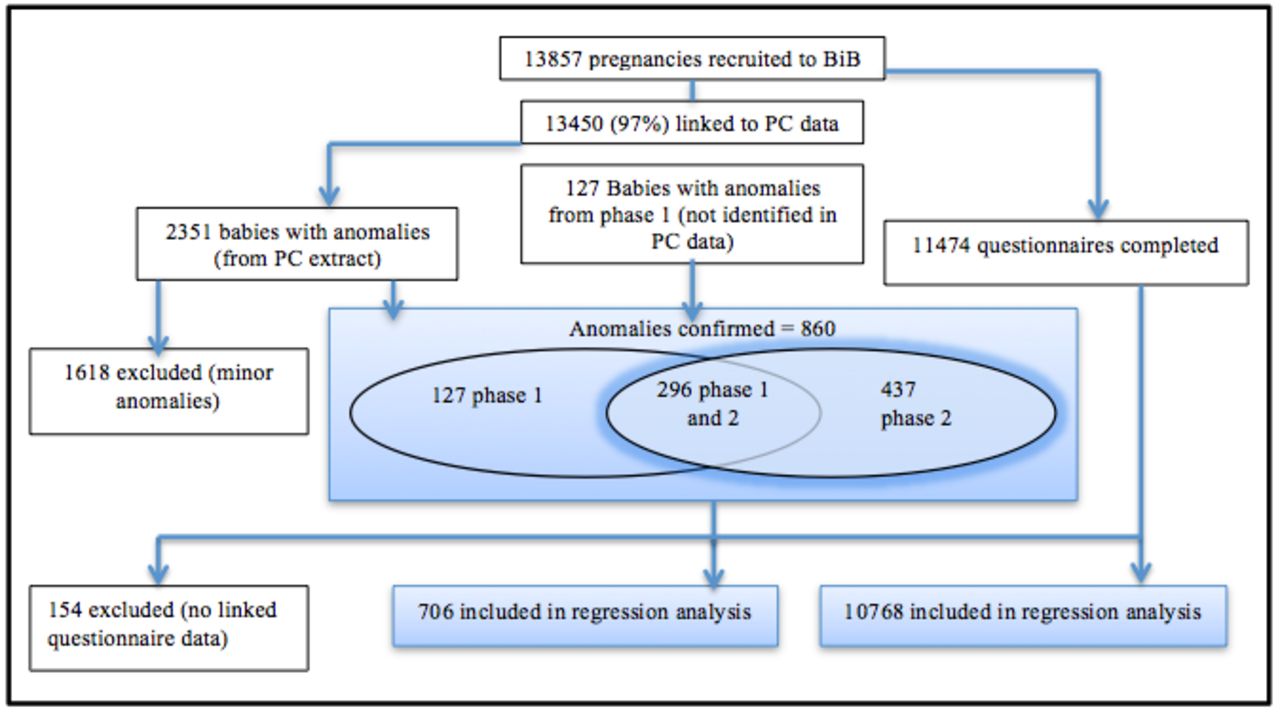
Flow diagram of steps in the analysis. BiB, Born in Bradford; PC, primary care.
Risk factors
We reviewed the following maternal risk factors for CA: ethnic origin (White British, Pakistani and Other); age of mother (<20, 20–34 and >34 years); educational attainment (less than five General Certificate of Secondary Education (GCSE) equivalents; five or more GCSE equivalents at grades A–C; two Advanced Level equivalents; diploma, degree or higher degrees; other; unknown; and foreign unknown); socioeconomic status (Index of Multiple Deprivation 2010 (IMD)22); smoking (whether mother smoked during pregnancy or not); alcohol consumption (drank alcohol during pregnancy or 3 months before pregnancy (yes or no)); and consanguinity (first cousin, second cousin, other blood relation (less than second cousin) or non-consanguineous). We categorised results for body mass index (BMI) and oral glucose tolerance test in accordance with WHO guidelines.23 24
Statistical analysis
For risk factors, we estimated univariate risk ratios (RRs) and 95% CIs for the occurrence of an anomaly with Poisson regression and robust error variance. We calculated risks for all ethnic groups and separately for White British, Pakistani and other groups. The CIs for smoking and BMI included one in univariate analyses and so were not included in multivariable analyses. We performed a test for interaction between consanguinity and IMD score. To address issues of multiple testing, all models were rerun using 99.9% CIs (data not shown). All analyses were performed in Stata (version 13).
Results
An additional 437 children with a CA were identified using the primary care database. Comparison of rates with BINOCAR was based on 1408 CAs noted in the total of 860 children remaining after the exclusion of minor CA. Table 1 compares the prevalence of anomalies in Bradford with National data reported by BINOCAR.2 In 2014, BINOCAR reported a prevalence (based on 2012 data) for CA, excluding chromosomal disorders, of 184 per 10 000 live births (table 1).2 Up to age 1 year, we report a total CA rate of 376 per 10 000 live births and a rate of 571.6 per 10 000 live births, including cases ascertained in all children under 5 years. Both these rates specifically exclude children with chromosomal or metabolic disorders. This difference in exclusions is due to BINOCAR registering children with metabolic disorders only if they also have a structural anomaly. BiB includes children with metabolic disorders whether or not they had a structural anomaly. Despite the large difference in BINOCAR rates at age 1 year and BiB rates at age 1 year, the phase 1 study10 found a similar 305.74 per 10 000 live births, helping to explain the influence of the Bradford demographics on the high numbers, before additional cases are added post age 1 year using primary care records. Regression analyses were based on 706 children with CA, including metabolic and chromosomal conditions for whom BiB questionnaire data were available. The comparison group was the 10 768 cases without CA but for whom questionnaire data were available.
Comparison of CA prevalence per 10 000 live births comparing BiB and BINOCAR data
Table 2 shows the characteristics of mothers in the BiB study who gave birth to children with or without CA, combining cases from phases 1 and 2. Table 3 shows the univariate and multivariable analyses of the risk factors included in table 2. The cohort was multiethnic, with 40% White British, 45% Pakistani and 15% reporting different ethnicities we refer to as ‘Other’ (table 2). In keeping with rates from phase 1,10 the proportion of children with anomalies born to Pakistani mothers was higher than those born to mothers in the cohort overall (53% for Pakistani children and 47% for White British and Other combined). Figure 2 demonstrates the age of the child when they received their first CA diagnoses. Without the additional cases from primary care data, this plot would only show diagnoses up to age 1 year, a total of 600 children. Primary care data add a further 260 cases (30%).
Demographics, lifestyle and clinical information of Born in Bradford mothers who gave birth to children with or without a CA by ethnic group

{kind=link}
{kind=link}
Total number of CA diagnoses per year, and additional CA diagnoses per year using phase 1 and phase 2 data combined.
Less than 1% of children of White British origin with CA were the offspring of first cousin unions compared with 49% of children with CA in the first cousin Pakistani subgroup. There was a positive stepwise association between the CA prevalence and the degree of consanguinity in the Pakistani subgroup: 9.5% of first cousin progeny, 7.7% of second cousin progeny, 7.5% of beyond second cousins and 4.8% in non-consanguineous progeny. Sixty-six per cent of the BiB cohort who had completed questionnaires lived in areas defined by the IMD as the most deprived fifth of England (table 2). The adjusted rates show an excess risk to children born to mothers in the least deprived fifth overall, which was also shown in phase 1.10 However, the numbers are very small and thus should be treated with caution (table 3). Consanguinity was found to be a major risk factor for CA in Pakistani mothers in both adjusted and unadjusted rates (multivariable RR 1.87, 95% CI 1.46 to 2.38) and maternal age >34 years for mothers of other ethnicities (multivariable RR 2.19, 95% CI 1.36 to 3.54). Conversely, a higher level of education was associated with a low risk of CA (multivariable RR 0.78, 95% CI 0.62 to 0.98) (table 3).
Univariate and multivariable RRs of CAs related to demographic, lifestyle and clinical risk factors in the Born in Bradford cohort by ethnic group
Discussion
In the most recent BINOCAR report of data for 2012, less than 2% of live births were diagnosed after 1 year of age.2 Using primary care data on children aged 0–<5 years revealed an additional 437 children with CAs, almost two times of those previously reported in phase 1.10 Only 70% of diagnoses in the present study were made prior to the children’s first birthday. Similar results have been reported by others and confirm the value of primary care data as a source to investigate CA.5 12–17 One review of CA reported in a UK database of primary care records (The Health Improvement Network) revealed that 72% of cases were diagnosed up to age 1 year, and after including diagnoses made up to age 5 years, rates increased from 198 per 10 000 to 277 per 10 000.12 The overall prevalence of heart defects was reported as lower for infants diagnosed up to age 1 year than infants diagnosed up to age 6 years in a further primary care database study.14 Late detection of heart CA could be attributable to some cases being missed at antenatal screening due to detection being difficult.25 Two studies using the GPRD found an increase in CA diagnoses when post-age 1 year CA diagnoses were included.14 17 Excluding chromosomal and metabolic conditions and considering diagnoses for age 0–<5 years, we found the profile of disorders in terms of bodily system categories to be consistent with those reported previously.2 10 12 26 The only exception is nervous system disorders, seen as the most common group in our study and in CA research specific to the UK.2 10 Considering the percentage increase by bodily system group, skeletal dysplasias increased considerably from age 1 year to age 5 years (210%), primarily due to diagnoses of short stature, followed by nervous system (77%), due to an inflation of hearing loss in Bradford, and respiratory (44%) disorders that are known to be high in Bradford27 (table 1). Some of the conditions in these subgroups are not expected to be detected in the prenatal period,8 but our data suggest that they may be taking longer to diagnose than previously thought, which has significant clinical implications. Delayed diagnoses are seen to create increased complications with care coordination and create a reliance on emergency care.28–31 We also assessed the effect of improved ascertainment on the point estimates and statistical significance of the risk factors for CA. We found no substantial change in these risk factors, even with a slightly different CA profile. Changes to statistical significance of risk factors would have had implications for comparative analyses between registries with different ascertainment methods.
Our findings, combined with other primary care database studies for CA ascertainment, therefore suggest that there are more than 2% of CA diagnoses being made after age 1 year, and registries may need to be more specific about their data collection methods for later diagnoses. In England, the recently established National Congenital Anomaly and Rare Disease Registration Service (NCARDRS)32 specify that they collect data, including risk factors, via notifications after age 1 year. The service is new, and the longer-term picture in terms of comprehensive ascertainment is not yet known. Our study demonstrates that ascertainment is strengthened with the use of routine NHS data sources. Consequently, the increased prevalence of children living with CAs in the community may require additional specialist resources for paediatric, obstetric and genetic care for this, as-yet, underascertained cohort. The results could also have implications for transition to adult services.
There are limitations to our study. The time of diagnoses based on Read code entry into primary care systems has been reported as later, on average, than dates of actual diagnosis by general practitioners.17 The cross mapping of Read to ICD-10 is vulnerable to discrepancies due to multiple Read codes matching one ICD-10 code. We accounted for this by performing a clinical review, assigning the most appropriate Read code to ICD-10 match. CA cases that are stillborn or diagnosed antenatally, resulting in termination, are not well recorded in women’s primary care records, thus underestimating CA cases. NCARDRS highlights that 71% of CA are detected antenatally, and 42% of CA diagnosed antenatally resulted in termination.32 Another CA study found fetuses diagnosed with a major CA had a high likelihood of termination of pregnancy, at 50% for consanguineous unions and at 60.9% for non-consanguineous pregnancies.33 BiB does not report terminations of pregnancy or miscarriages because recruitment is at 26–28 weeks gestation. This current, unpublished work on the BiB cohort has revealed a high level of concordance between self-report of consanguineous marriage and genetic relatedness. There is also a suggestion that the level of genetic relatedness exceeds that expected from self-report, and this may be a function of traditional male socio-occupational groupings. These aspects are being investigated in the BiB cohort as part of an ongoing programme of research.34
Conclusion
We have combined personal and clinical information from a large cohort study with routine primary care data to produce a more comprehensive assessment of the burden of CA in live births. We have demonstrated that more complete case ascertainment of CA can be achieved by linking to primary care data, and by using these, data we are able to detect later diagnoses up to the age of 5 years. Our study also reaffirms consanguinity as a major risk factor for CA in the Bradford Pakistani community.
Acknowledgments
This paper presents independent research by a PhD candidate. The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research or the Department of Health. We thank the families who took part in the Born in Bradford study, the midwives for their help in recruitment, the paediatricians and health visitors, the Born in Bradford team, which included interviewers, data managers, laboratory staff, clerical workers, research scientists, volunteers and managers.
References
Footnotes
Contributors CB, RP, JW and NS conceived the idea and designed the protocol, with advice from DM on primary care database linkage and conversion of Read codes. ES, RP and PC reviewed all anomalies reported. CB did the statistical analysis, which was overseen by RP with additional interpretation by AHB. All authors contributed to and have approved the final analyses.
Funding This work was supported by a Bradford University studentship, in conjunction with the White Rose Consortium, and the National Institute for Health Research, Collaboration for Leadership in Applied Health Research and Care Yorkshire and Humber programme ‘Healthy Children Healthy Families Theme’ (IS-CLA-0113–10020). The sponsors of this study had no role in the study design, data collection, analysis or interpretation or writing of the report.
Competing interests None declared.
Ethics approval Ethics approval for the cohort study was provided by Bradford Local Research Ethics Committee (reference 06/Q1202/48).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Additional information is available on request from the corresponding author c.bishop1@bradford.ac.uk.