Article Text

Abstract
Background Rett syndrome (RTT) is a severe, progressive neurodevelopmental disorder with multisystem comorbidities that evolve across a patient’s lifespan requiring attentive coordination of subspecialty care by primary care providers. A comprehensive, up-to-date synthesis of medical comorbidities in RTT would aid care coordination and anticipatory guidance efforts by healthcare providers. Our objective was to review and summarise published evidence regarding prevalence of RTT medical comorbidities across all relevant organ systems.
Methods Search of PubMed from January 2000 to July 2019 was performed using the search terms (Rett and MECP2 AND patient) OR (Rett and MECP2 AND cohort). Articles reporting the prevalence of clinical findings in RTT were assessed with respect to the size and nature of the cohorts interrogated and their relevance to clinical care.
Results After review of over 800 records, the multisystem comorbidities of RTT were summarised quantitatively from 18 records comprising both retrospective and prospective cohorts (31–983 subjects). Neurological comorbidities had the highest prevalence, occurring in nearly all individuals with gastrointestinal and orthopaedic concerns almost as prevalent as neurological. With the exception of low bone mineral content which was relatively common, endocrine comorbidities were seen in only around one-third of patients. Although more prevalent compared with the general population, cardiac conduction abnormalities were the least common comorbidity in RTT.
Conclusions Effective care coordination for RTT requires knowledge of and attention to multiple comorbidities across multiple unrelated organ systems. Many issues common to RTT can potentially be managed by a primary care provider but the need for sub-specialist referral can be anticipated. Since the median life expectancy extends into the sixth decade with evolving subspecialty requirements throughout this time, paediatric providers may be tasked with continued coordination of these comorbidities or transitioning to adult medicine and specialists with experience managing individuals with complex medical needs.
- neurology
- rehabilitation
- gastroenterology
- genetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is known about the subject?
Rett syndrome (RTT) is a rare, genetic neurodevelopmental disorder with multiple non-neurological comorbidities that evolve with age. Healthcare providers play a key role in recognising, treating and directing care of comorbidities of patients with RTT. Observational cohort studies conducted over the past three decades have yielded information regarding prevalence of individual comorbidities.
What this study adds?
In this review, neurological comorbidities are highly prevalent, occurring in nearly all patients. These are closely followed in prevalence by gastrointestinal and orthopaedic issues. Endocrine and cardiac comorbidities are less common though still more prevalent than the general population. Complex subspecialty requirements are anticipated for RTT based on this information.
Introduction
Classic (typical) Rett syndrome (RTT)1 is a severe, progressive neurodevelopmental disorder occurring in approximately 1 in 10 000 female births.2 There is an estimated worldwide prevalence of between 1 in 20 000 and 40 000 people making2 it one of the most common genetic causes of developmental and intellectual impairment in females.3 The syndrome is characterised by an initial period of normal development followed by psychomotor regression, deceleration of head growth and development of distinctive repetitive, purposeless hand movements.4–6 Due to symptom evolution over time, RTT is considered a progressive disorder but, contrary to a long-held misconception, it is not a neurodegenerative condition.7 RTT is caused by >300 distinct loss-of-function mutations in the gene MECP2 (methyl-CpG binding protein-2) on the X-chromosome.8 The MECP2 protein is an essential transcriptional regulator in the brain required for normal neurodevelopment.9 Due to major strides in understanding the molecular pathogenesis over the past three decades and successful reversal of symptoms in mouse models,10 11 there is optimism for disease modifying therapies in the future.
Despite the present inability to modify the course of the disorder at a mechanistic level, over two decades of research on the natural history of RTT1 has shown us that with appropriate symptom-directed care, people with RTT can live long and meaningful lives. Compared with an estimate of survival using historical data from the earliest published cohort of people with RTT which found very low rates of survival beyond the third decade of life,12 analyses of modern cohorts demonstrate markedly improved survival as symptom management has evolved13 with the most recent report from a large North American longitudinal cohort finding 70% of individuals with typical RTT surviving to at least 50 years of age.5 14 The multisystem involvement of the disorder with comorbidities that evolve throughout a patient’s lifespan thus present a significant challenge to the physicians tasked with effectively coordinating the management of these symptoms which will often require subspecialty consultation.
While there have been many publications over the past 20 years addressing individual organ system comorbidities in RTT, a comprehensive synthesis in accordance with the multisystem involvement of the disorder would provide a global understanding of RTT and better facilitate the daunting challenge of effective care coordination. To address this challenge, we performed a review of published literature regarding RTT symptomatology with the goal of delineating the prevalence of comorbidities across all organ systems.
Methods
Database search
In July 2019 we performed a search of PubMed (from 2000) using the search terms (Rett and MECP2 AND patient) OR (Rett and MECP2 AND cohort). Due to the anticipated low number of articles addressing symptom prevalence, we implemented a broad initial search strategy to minimise the likelihood of excluding relevant articles. Full details of the literature search strategy are described in the Preferred Reporting Items for Systematic Reviews and Meta-Analyses flow diagram presented in figure 1.
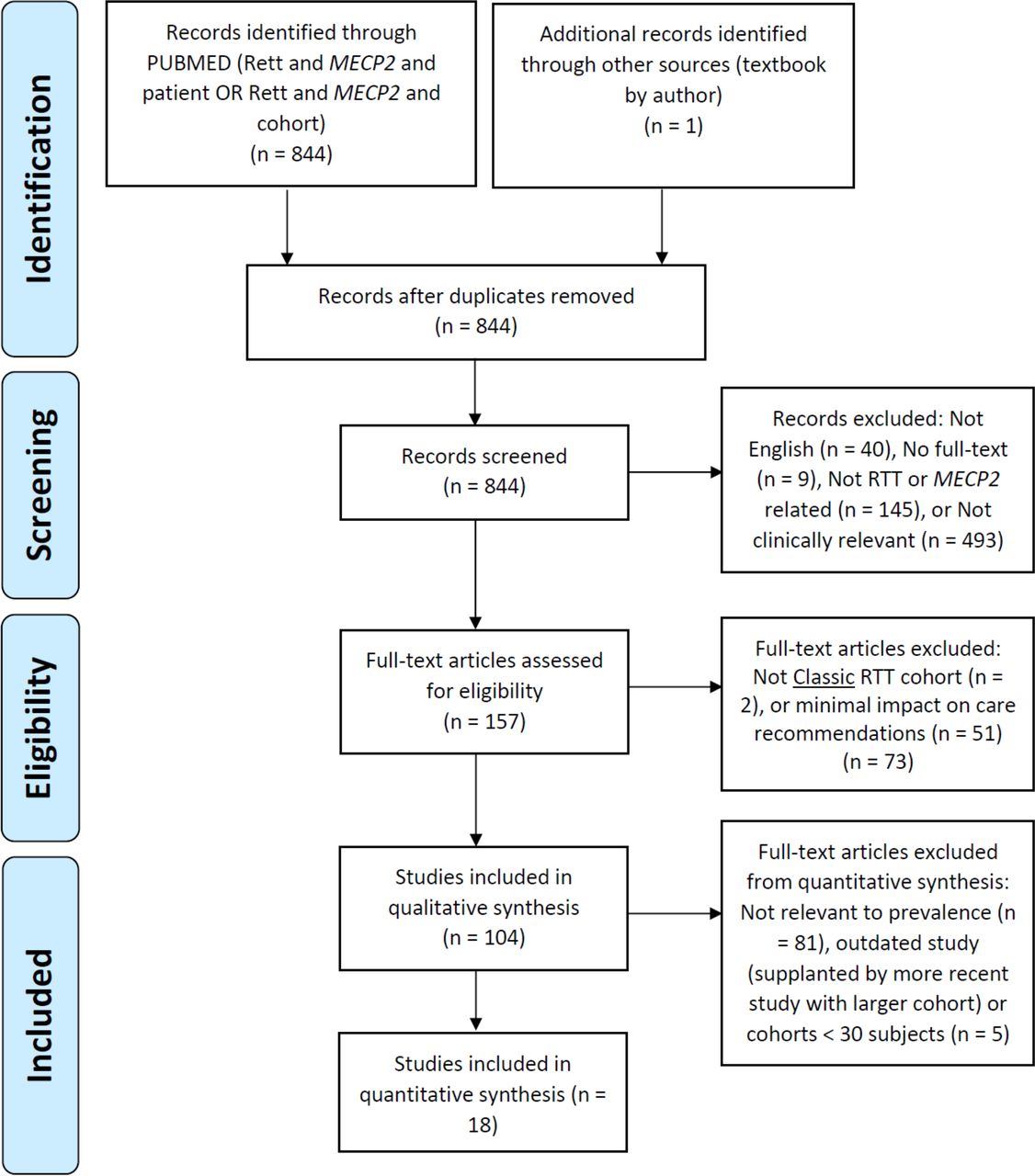
{kind=link}
Preferred Reporting Items for Systematic Reviews and Meta-Analyses flow diagram. RTT, Rett syndrome.
Article selection
We defined the inclusion and exclusion criteria for final article selection with input from patient advocacy group representatives. From the initial search criteria we first excluded articles that were not in English (n=40), did not have full-text access (n=9), focused solely on non-clinical topics (n=493) or were related to disorders other than RTT (n=145). Next, from the resulting full-text articles (n=157) we included only articles reporting on classic RTT1 containing data relevant to clinical care (n=104). We worked as a group to select articles based on the inclusion criteria with the group leader (TAB) moderating the process. The selection process comprised two stages: an initial stage involving title and abstract review and second stage involving the full-text. All authors participated at both stages. We used the final list of 104 articles for a qualitative synthesis informing general clinical guidance (‘Consensus guidelines on managing Rett Syndrome across the lifespan’ (Fu C, et al. BMJ Paediatrics Open 2020, in press)). For the final quantitative synthesis we only included articles (n=18) specifically describing prevalence of clinically relevant comorbidities in cohorts of ≥30 subjects that had not been superseded by a more recent publication studying the same comorbidity in a larger (at least one order of magnitude) cohort (‘outdated’).
Patient advocacy group involvement
Patient advocacy groups (International Rett Syndrome Foundation and Rett Syndrome Research Trust), represented by parents of individuals with RTT, provided feedback on the relevance of the literature review. They will help disseminate these findings as part of ‘Consensus guidelines on managing Rett Syndrome across the lifespan’ (manuscript accepted for publication).
Results
Our search strategy (figure 1) yielded 844 citations that were screened further by title and abstract. Of these, 687 articles were excluded, leaving 157 full-text articles for more detailed review. From the detailed review 104 articles were retained for a qualitative synthesis of clinical care guidelines in RTT (manuscript in preparation). These articles originated from countries throughout the world including USA, Australia, Canada, UK, Italy, Austria, The Netherlands, Sweden, Turkey, Portugal and Israel. Most (79%) of the articles involved original research studies or review articles that did not specifically address prevalence of non-neurological comorbidity but were informative for clinical management and there were a small number (5%) of articles that reported prevalence data from small prospective cohorts. Importantly, 18 articles (17%) specifically quantified the prevalence of clinically relevant comorbidities in large cohorts of patients with classic RTT. These articles were then used for the quantitative synthesis regarding prevalence of medical comorbidities (table 1).
Prevalence of RTT comorbidities
The characteristics of the 18 included studies are presented in table 2. Most of the articles were from North America: 11 articles from the USA and 1 from Canada; additionally, there were 4 articles from Australia, 1 from Portugal and 1 from Italy. The cohort size ranged from 31 to 983 subjects. The study designs were divided equally between prospective (9 of 18 articles) and cross-sectional assessments (9 of 18 articles). Most of the prospective studies (8 articles) involved in person assessments; 5 studies with multisite data collection15–19 and 3 studies with single institution cohorts.20–22 A single prospective study used a national patient registry capturing data from caregiver completed questionnaires.23 The majority of cross-sectional studies (5 articles) also involved data collection through in person assessments; 2 studies that were multisite24 25 and 3 that were single institution.26–28 Of the remaining cross-sectional studies (4 articles), three involved data collection through remotely disseminated caregiver completed questionnaires29–31 and a single study involved retrospective medical records review.32
Description of articles included in the quantitative synthesis of Rett syndrome comorbidities
The multisystem nature of the disorder is quite apparent based on the observational studies identified in this scoping review (table 2). As expected for a neurodevelopmental disorder, neurological concerns are the most prevalent of all comorbidities (table 1) including autonomic dysregulation, epilepsy, swallowing dysfunction, sleep dysfunction, abnormal movements and behaviour disturbances. The autonomic dysregulation manifests most prominently as an irregular breathing pattern which occurs in nearly 100% of individuals with RTT over the lifespan with the majority presenting by age four with varying levels of rapid breathing, breath-holding and/or air-swallowing that fluctuate in intensity while awake and disappear during sleep.16 Epilepsy is nearly as prevalent as breathing irregularity with nearly 90% of individuals receiving this diagnosis across the lifespan, the majority by 6 years of age.17 Impaired swallowing function has equal prevalence to epilepsy over the lifespan but becomes apparent at a younger age often manifesting in early childhood as prolonged feeding times, choking/gagging on food and/or aspiration of liquids.29 Sleep dysfunction was another highly prevalent neurological comorbidity reported in around 90% of individuals manifesting as difficulty falling asleep and/or staying asleep with important impacts on overall family quality of life.23 31 Behaviour disturbances occur in nearly all individuals with RTT across the lifespan with internalising behaviours such as anxiety or mood swings being much more common overall than externalising behaviours such as aggression, hyperactivity or self-injury which are seen in about 40% of individuals, almost always with onset before 3 years of age and which improve over time in contrast to the internalising behaviours.15 Outside of the repetitive hand movements that are part of the main diagnostic criteria of typical RTT, additional movement abnormalities are found in 60%–80% involving varying combinations of dystonia, tremor, myoclonus, chorea and progressive limb rigidity with bradykinesia resembling parkinsonism.20–22
In addition to the highly prevalent neurodevelopmental issues, gastrointestinal and orthopaedic comorbidities are both also quite prevalent, each reported in over 80% of individuals.29 30 The gastrointestinal issues primarily involve constipation but around 40% also have gastro-oesophageal reflux. Both symptoms are potentially related to underlying autonomic nervous system abnormalities causing impaired gastrointestinal motility. Poor growth is reported in around 40% of individuals primarily involving impaired weight gain and/or linear growth deficits.29 The impaired growth is thought mostly secondary to chewing/swallowing dysfunction causing inadequate caloric intake because improvements in height and weight gain are seen after calorie supplementation via gastrostomy tube.33 Biliary tract disease does not appear to impact individuals with RTT at a higher rate (4%) than the general population but may be difficult to recognise due to non-specific symptoms overlapping with behaviour disturbances and the underlying communication impairments in RTT.30 Scoliosis is the most frequently encountered orthopaedic issue with 85% of individuals 16 years or older diagnosed with this finding. Its occurrence is age-dependent though with much lower prevalence in childhood and increasing prevalence through adolescence.19 Additional important orthopaedic complications include hip displacement (either subluxation or dislocation) that may be seen in a little over half of individuals32 and also a three to four fold higher prevalence of bone fractures compared with typically developing individuals.24 28 The high prevalence of fractures is potentially linked to low bone mineral content in RTT which is the most common endocrine comorbidity that is seen in over half of individuals beginning in childhood.28 The timing of secondary sexual characteristics is also frequently altered in RTT with early adrenarche or thelarche in a quarter and just under 20% with delayed menarche.18 Above normal levels of thyroid hormone are also found in about 20% but the clinical implications of this are unclear.27 Finally, cardiac conduction abnormalities involving prolonged QT interval are seen in 10%–18%.25 26
Discussion
Based on our approach with the literature to date, the comprehensive list of clinically relevant comorbidities in RTT is considered to be complete, especially due to the experiences with larger cohorts. There are some limitations with our approach to estimate the prevalence of these comorbidities. While we have selected articles whenever possible based on the best available evidence (level 3) involving medium to large cohorts with prospective data collection, there is the potential for bias because many of the studies analysed were generated from the authors’ own experience and approach with the largest cohort via the National Institutes of Health-funded Natural History Study of Rett and related disorders (National Health Service, U54 HD061222; ClinicalTrials.gov: NCT00299312/NCT02738281). This study required in-person assessments for data collection and as such potentially subject to ascertainment bias, however, generalisability is supported by similar prevalence findings when the same comorbidity was reported in different and unique cohorts with population-based sampling.5 13 The approach included studies with at least 30 subjects whenever larger cohorts were not available for a comorbidity. These smaller studies may miss the true prevalence of specific RTT comorbidities. Recruiting large cohorts is always a challenge in rare disease research and the recruitment of subjects with extreme symptoms and medically fragile conditions may be more difficult outside of a clinical setting. The smallest cohorts included in this synthesis were ascertained from clinical settings.20–22 27 28 32 These cohorts, though smaller, may reflect a fuller spectrum of disease severity.
While it is possible that some of the prevalence estimates of medically relevant RTT comorbidities require further investigation to be reliably generalisable, the available information supports the medically complex nature of this disorder and the importance of coordinating care across multiple specialties. Until additional features are better resolved through longer observations in large cohorts, especially in those that include the older and most medically fragile individuals with RTT, medical features outside this list must be considered to not be comorbid with RTT and are deserving of further medical investigations to determine their aetiology.
In conclusion, RTT is a medically complex neurodevelopmental disorder impacting multiple organ systems in an evolving fashion from childhood through the sixth decade of adulthood. With the advances in healthcare and technology, improved and earlier genetic testing, robust research in RTT, and active patient advocacy from families and clinicians, individuals with RTT are surviving well into adulthood while living healthier and more meaningful lives. Primary care providers are uniquely positioned to most effectively manage the individual and family by drawing on the accumulating knowledge regarding the natural history of the disorder to anticipate and coordinate the complex multidisciplinary requirements. When children with RTT grow into adulthood, paediatricians may be tasked with continued care coordination for these comorbidities or transitioning primary care to adult medicine specialists with experience managing individuals with complex medical needs.
Acknowledgments
We thank the RTT patient advisory groups (International Rett Syndrome Foundation and Rett Syndrome Research Trust) for their input and assistance in dissemination of our findings. We sincerely thank all of the individuals and families that have participated in this research. Thanks to Dr Walter Kaufmann for comments on an earlier stage of this manuscript.
References
Footnotes
Contributors CF, JN, AP and TB conceptualised and designed the literature search. CF, DA, DL, EM and RW initiated the search and a first draft. All authors contributed to subsequent drafts. TB, as group leader, supervised and moderated the search, initial drafts, the overall collation of the figure, tables and final manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Funding CF: International Rett Syndrome Foundation; DA: Rett Syndrome Research Trust; EM: NIH U54 HD061222, Rett Syndrome Research Trust; RW: Rett Syndrome Research Trust; DL: Rett Syndrome Research Trust, NIH U54 HD061222; JL: NIH U54 HD061222, Rett Syndrome Research Trust, International Rett Syndrome Foundation; KM: International Rett Syndrome Foundation; Bernhard Suter: Blue Bird Circle, NIH U54 HD061222; DG: Blue Bird Circle, NIH U54 HD061222; JN: NIH U54 HD061222, U54 HD461222, Rett Syndrome Research Trust; AP: NIH U54 HD061222, Rett Syndrome Research Trust; TB: International Rett Syndrome Foundation, Rett Syndrome Research Trust, NIH U54 HD061222, Children’s Hospital Colorado Foundation Ponzio Family Chair in Neurology Research.
Competing interests EM: funding from the NIH and International Rett Syndrome Foundation; clinical trials with GW Pharmaceuticals, Zogenix, Marinus; Consultancy to Stoke therapeutics. DL: consultancy for AveXis, clinical trials with Acadia, Anavex, GW Pharmaceuticals,. SS: speaker Bureau for GW Pharmaceuticals. BS: funding from the NIH and Blue Bird Circle; clinical trials with Acadia. DG: funding from the NIH and Blue Bird Circle; clinical trials with GW Pharmaceuticals, Acadia, Anavex, Newron; consultancy for Acadia and Trend Community Pharmaceuticals. JN: funding from the NIH; consultancy with Acadia, AveXis, Biohaven, GW Pharmaceuticals, Kurro, Neuren, Newron, Ovid, Takeda and Teva. JL: funding from NIH; consultancy from International Rett Syndrome Foundation and GW Pharmaceuticals. AP: funding from the NIH; consultancy for Anavex, AveXIs, Acadia and GW Pharmaceuticals; clinical Trials with Anavex, Acadia, GW Pharmaceuticals, and RSRT; TB: funding from the NIH, International Foundation for CDKL5 Research and Loulou Foundation. Consultancy for AveXis, Ovid, GW Pharmaceuticals, International Rett Syndrome Foundation, Takeda and Marinus; Clinical Trials with Acadia, Ovid, GW Pharmaceuticals, Marinus and RSRT; all remuneration has been made to his department.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available.