Article Text

Abstract
Objective The objective of this study is to study familial inheritance for Blount disease to create better understanding of the aetiology of Blount disease.
Methods After reviewing patient files and conventional roentgenologic imaging, 139 patients with Blount disease were included in this cross-sectional study, of which 102 patients were interviewed. During the interviews, patient characteristics and family history were collected. Blood samples were taken from five patients and three families and a whole exome sequencing was performed.
Results Although patients came from all over the country, 90% of the patients belonged to the Akan tribe. A positive family history was found in 63 families (62%), of which, almost two-third had a positive family history in a first-degree family member. In most of the cases (64%), the varus legs resolved over time. In 9%, severe bowing remained ‘just like the patient’. The results of the whole exome sequencing did not show a genetic predisposition.
Conclusion This study describes a large group of Blount patients. Because of the high numbers of positive family history and the centralisation of patients in the Akan region, a familial predisposition is suggested. Further genetic research is essential for better understanding of the possible multifactorial aetiology in Blount disease.
- genetics
- growth
Data availability statement
Data are available upon reasonable request at the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is known about the subject?
Blount disease is a rare paediatric orthopaedic disease, which results in genu varum, internal rotation and procurvatum.
The best-found hypothesis is a combination of factors such as obesity, early walking age, pre-existing varus and race, also called ‘increased mechanical force hypothesis’.
What this study adds?
Detailed family history in a large group of patients with Blount disease.
Large study in a sub-Saharan African country, until now most studies on the aetiology of Blount disease are conducted in high income countries.
Two-third of the patients in the early onset group reported a positive family history for varus legs.
Introduction
Blount disease is characterised by a growth arrest and disturbed endochondral ossification of the posteromedial part of the proximal tibia. This results in genu varum, internal rotation and procurvatum.1–3 Three different forms of the condition are described, based on the age of onset of the condition. Infantile, or early onset, starts before the age of 4 years, juvenile onset starts between the age of 4 and 10 years and adolescent or late onset starts after the age of 10 years.2 4
The aetiology of Blount disease is not well known. The best-found hypothesis is possibly a combination of factors such as obesity, early walking age, pre-existing varus and race.4–10 Although there are several hypotheses, most articles support the hypothesis of the combination of factors, often referred to as the ‘increased mechanical force hypothesis’.
Most studies supporting the ‘increased mechanical force hypothesis’ are conducted in high income countries, mostly the USA. The prevalence of overweight and obesity in the American population is much higher, respectively, 32% and 17%, compared with the prevalence in sub-Saharan Africa, respectively, 11% and 3%.11 12 This stands in contrast to earlier published work showing a strong relation between obesity and developing Blount disease in developed countries,6 8 13 and the relative high prevalence in the African population.9 14 15 Also in the USA, Blount disease was found more prevalent in the Afro-American ethnic group compared with the Caucasian ethnic group.4 6 7 16–20 It is suggested that Blount disease is more common in the African and Afro-American population because of the earlier walking age and the greater laxity of the knee ligaments compared with the Caucasian population.15 18 But this does not explain why relatively large groups of patients with Blount disease are found in Finland and Japan.21 22 In addition, a genetic predisposition is suggested.9 10 23
Most studies describing familial occurrence of Blount disease are case reports or retrospective studies in which affected siblings were an incidental finding.10 22 24–27 The only studies which actively conducted a family history found a positive family history in 14% and 45% of the early onset Blount patients,21 28 and a negative family history for all adolescent Blount patients.19 21
To the best of our knowledge, no studies focusing at genetic predisposition are conducted among patients with Blount disease.
In this study, we investigated the familial influences and possible familial predisposition in Blount patients. It is hypothesised that Blount disease is seen more often in children with a positive family history of Blount disease or varus legs compared with those with a negative family history and that there might be a familial predisposition.
Methods
Patient files and conventional roentgenologic images from patients diagnosed with Blount disease seen in a mission hospital in rural Ghana between May 2010 and March 2018 were reviewed. In total, 206 patients with the diagnosis of Blount disease were selected. All patient files and X-rays were evaluated by an orthopaedic surgeon and the first author. Exclusion was based on patients younger than 2 years of age at presentation, missing patient files, diagnosis not made by an orthopaedic surgeon and no X-ray available for evaluation, X-ray did not meet the Langenskiold classification for Blount disease.22 After exclusion, 139 cases of confirmed Blount disease were included. These patients were approached for an interview after informed consent was given. The interviews were conducted over the phone (n=74) or during a face-to-face meeting (n=28). In April 2018, 93 interviews were conducted by the first author. Because of the language barrier, the same translator was used for all the interviews. From nine patients, data conducted during interviews taken in December 2014 were used, because these patients were not reachable at the time of this research. Patients or the public were not involved in the design, or conduct, or reporting or dissemination plans of our research. The Ghanaian medical ethical committee has given ethical approval to perform this study (ref: CHRPE/AP/152/18).
Patient characteristics were collected through interviews (see online supplemental appendix 1). Tribes were allocated to one of the eight following ethnic groups: Akan, Mole-Dagbon, Ewe, Ga-Dangme, Gruma, Guan, Grusi or Mande, or assigned to ‘others’.
Supplemental material
Interviews were completed with one or both of the parents or the family member who raised the patient. In every case, the interviewee stated that there was a positive family history in the other side of the family, this needed to be confirmed by the other side of the family. A positive family history for bowed legs required to meet the following criteria; idiopathic varus deformity which started in childhood. Anamnestic cases of rachitic and polio were excluded. Self-reported family history has a sensitivity and specificity above 70% according to earlier studies in cancer and non-communicable diseases.29 30 Janssens et al30 noted that these numbers will be higher in if patients and family members are more aware of their condition. Blount disease is a visible condition and complicates daily mobilisation, this increases the awareness and therefore the accuracy of the self-reported family in Blount disease.
After the interviews were conducted, 13 families (patient and both parents) with the most comprehensive family history were selected for the genetic research in collaboration with the department of clinical genetics. From all 13 families, an extended family tree was made during a second interview, blood samples were taken and a second informed consent form was signed (online supplemental appendix 2).
Supplemental material
From every family, two Ethylene Diamine Tetra Acetic acid (EDTA) tubes with blood were obtained per person, DNA was isolated within 7 days. Based on the extended family trees obtained from the 13 families, five patients and three families (patient and both parents) were selected for diagnostic whole exome sequencing (WES) and variant calling was performed as described.31 Briefly, exome capturing was done using the Agilent SureSelectXT Human All Exon v5 library prep kit (Agilent Technologies, Santa Clara, CA, USA). Libraries were sequenced on an Illumina HiSeq 4000 instrument (Illumina, San Diego, CA, USA) with 101 bp paired-end reads at a median coverage of 75× at BGI Europe (BGI, Copenhagen, Denmark). Sequence reads were aligned to the hg19 reference genome using Burrows-Wheeler Alignment version 0.5.9-r16.14. Variants were called using the unified genotype Genome Analysis Toolkit, version 3.2-2 and annotated in a custom-built annotation pipeline developed for diagnostics. WES was both used to identify potential causal variants in known Mendelian disease genes in pedigrees, and also to generate massive variant data sets of the exomes from all pedigrees which could then be compared between samples for shared identical (likely) pathogenic variants or for non-identical variation in a communal candidate disease gene. Variant interpretation and classification were done adhering to professional guidelines.32
Continuous data are displayed as mean±SD and categorical variables are shown as a frequency and/or as a percentage.
Results
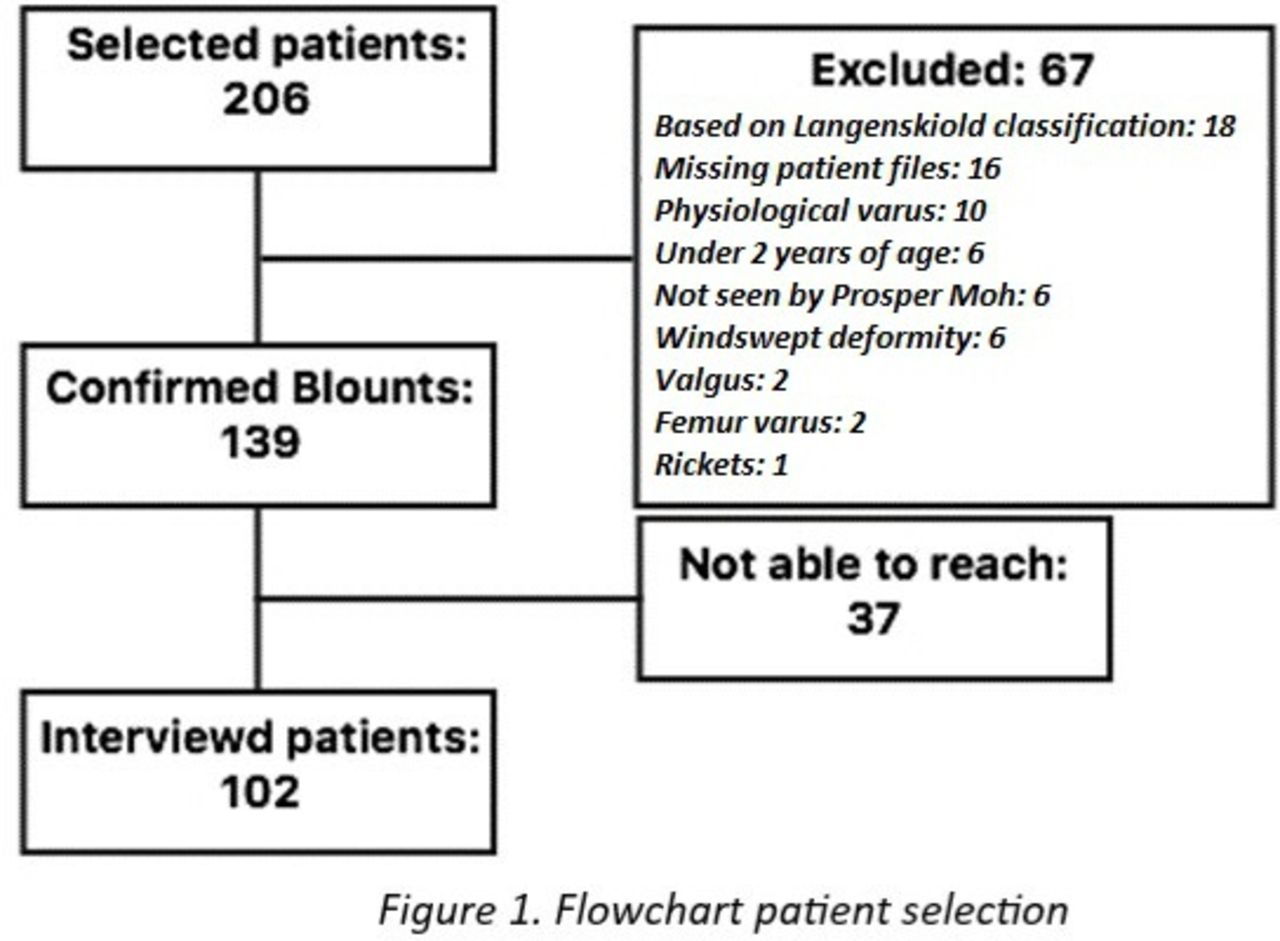
Out of 139 cases with confirmed Blount disease, 102 were included for analysis. All 37 excluded patients were not included because they were not reachable for an interview (figure 1). Data presented in this study concern the interviewed 102 patients, unless stated otherwise.

Flowchart patient selection.
Patient characteristics
Patient characteristics are displayed in table 1. Most patients were allocated to the early onset group (n=84), followed by the juvenile onset group (n=14) and the late onset group (n=4). The male female ratio was 1:4. In 43% of the cases, the condition was bilateral and in the unilateral cases, the left and right legs were almost as often affected (30% vs 27%). In our study population, 94% needed a surgical correction.
Patient characteristics
Patients came from all over Ghana, but analysing the distribution of the patients, most patients live in the Ashanti-region (55%), Western-region (19%) or Brong-Ahafo (17%) region and belong to the Akan tribe (88%). The Akan tribe is the biggest tribe in Ghana (48%) and consists of multiple subgroups. From the Akan patients of which we knew the subgroup (n=83), 59% was Asante (n=49). Interestingly, only 20% of all Ghanaian Akans belong to the Asante subgroup. Online supplemental appendix 3 shows a map with the distribution of patients in Ghana.
Supplemental material
Family history
A total of 137 cases of positive family history were found in 63 families. In the early onset group, two-third of the patients reported a positive family history, of which, almost two-third had a positive family history in a first-degree family member (35 out of 56). Furthermore, in the juvenile group and late onset group, there were families with a positive family history but not as frequent as in the early onset group.
Only two cases of positive family history were confirmed Blount disease, this concerned two brothers both operated in this hospital. A patient’s cousin with anamnestically the same disease was operated in a different hospital in Ghana. Nine other cases claimed to still have a severe, early onset, varus deformity similar to the patient, possibly having the same disease. As a result of the absence of diagnostics and treatment in these cases, the diagnosis remains unclear. Furthermore, 37 (27%) cases claimed to still have a mild varus deformity and in 88 cases (64%), the varus deformity resolved itself during childhood, at an average age of 7 years (diverging from 2 to 17 years).
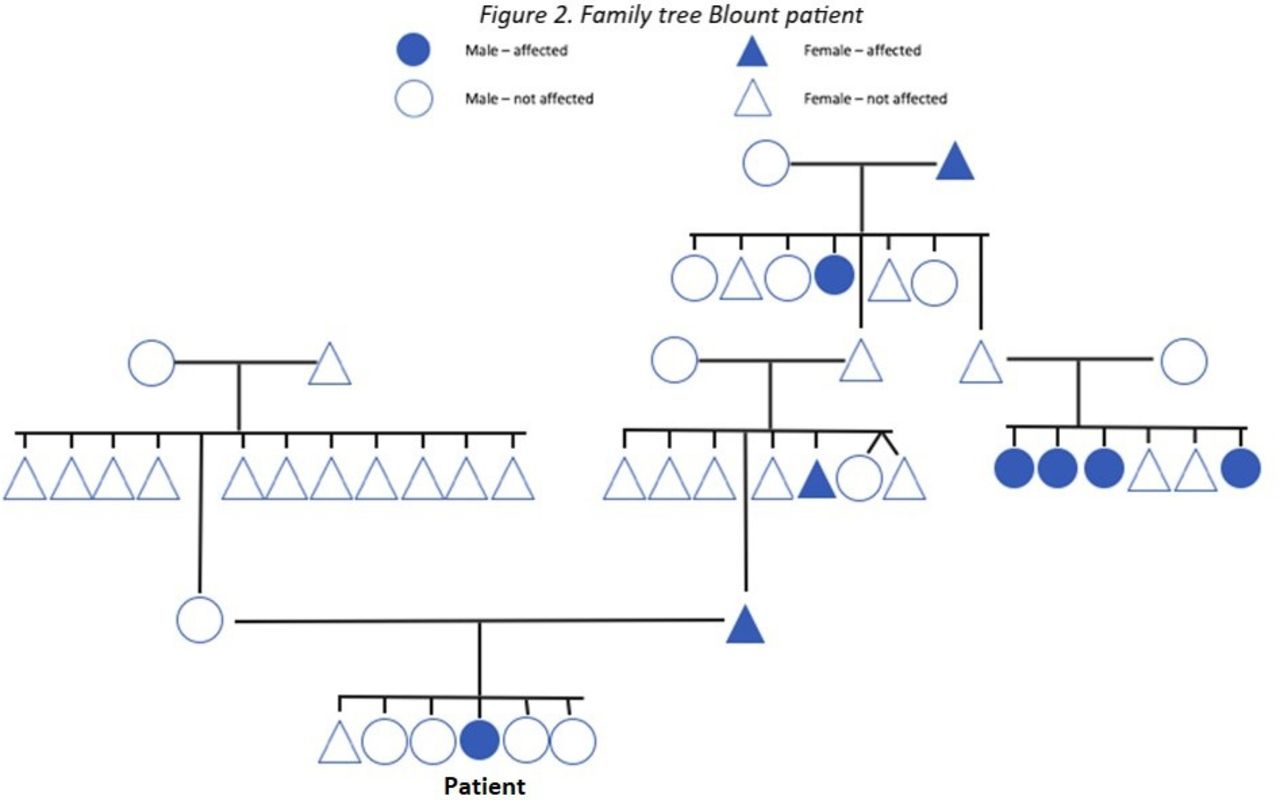
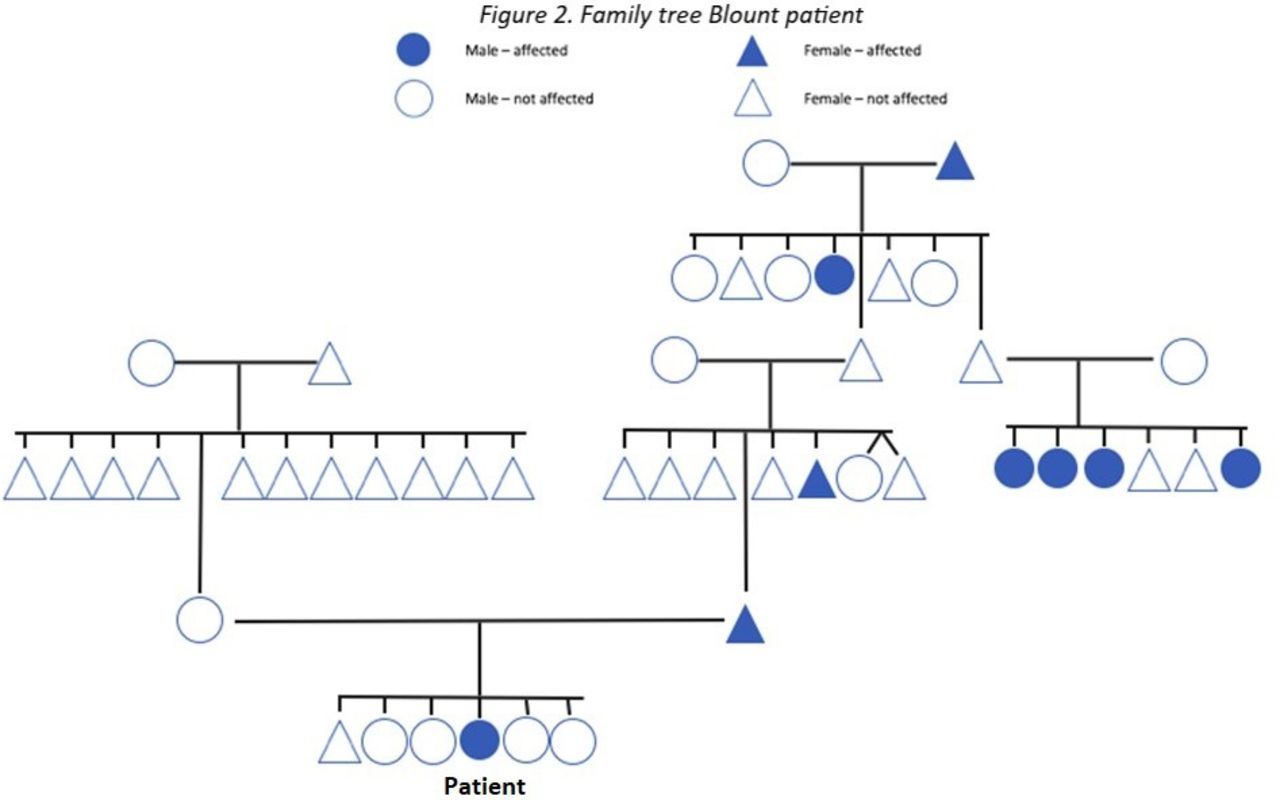
Anamnestically, one case of monozygotic twins and one case of monozygotic triplets were found in this study. All the twins and triplets’ siblings had varus legs from early childhood, however none of the siblings developed Blount disease. Table 2 shows the cases of positive family history and their severity. Figure 2 shows the family tree of one of the Blount disease patients.
Cases of positive family history
{kind=link}
{kind=link}
Family tree Blount patient.
Genetic predisposition
The first analysis of the WES did not show a clear genetic predisposition.
Routine diagnostic WES using genomic DNA of each patient was initially performed for defined gene panels consisting of genes associated with Mendelian-inherited disorders and Skeletal Dysplasia and/or Short Stature, including SHOX-gen (for gene panels content, see https://order.radboudumc.nl/en/genetics/rapid-exome-sequencing). In none of the patients did this result in the identification of a clinically relevant variant. Subsequently, the entire exome outside these gene panels was interrogated and compared between patients, in search of a biologically plausible candidate gene. This approach did not result in the identification of a candidate gene which could explain the presence of Blount disease in the study population.
Discussion
This study presents high prevalence of Blount patients, including 139 Blount patients in 8 years. Although, patients came from all over Ghana, 88% belong to the Akan tribe, nationwide less than 50% of the Ghanaians belong to the Akan tribe. Furthermore, in the early onset group, two-third of the patients reported a positive family history for varus legs. After analysing the interviews and literature, a relationship between the occurrence of Blount disease and familial inheritance for varus legs is suggested.
It is known that up to the age of 2 years, infants may have physiologic bowing. However, the family members with a positive family history for self-limiting varus in this study showed a relatively high age (average 7 years) for self-resolving the physiological bowing, which might suggest a more severe form of physiological varus even in the self-limiting group. In earlier bio mechanic studies, it was found that a 20° varus alignment in a 2-year-old child results in sufficient forces to reduce growth of the medial physis of the proximal tibia. For a 5-year-old child, the borderline force needed to inhibit the growth is a 10° varus alignment. These angles and forces are calculated in patients with a normal 50th percentile weight. Forces on the medial physis increase if patients are overweight, but weight has a weaker mechanical effect on the physis than the varus angle has.33 Our data are consistent with the hypothesis that Blount disease is primarily the result of the proximal tibial epiphysis responding to physical phenomena. Therefore, the high prevalence of familial inheritance for varus legs might explain the large population of Blount patients found in this study.
The contribution that familial inheritance for severe varus legs as an infant has on developing early Blount disease would also explain the higher number of positive family history in the early onset group compared with the juvenile group and late onset group, found in the study data and the current literature. In the juvenile case and late onset case, the bowing often started after gaining a lot of weight or trauma,4 20 22 34 which could also cause changing forces on the physis.
Little is known about the prevalence of Blount disease. In the literature, one to four cases of early onset Blount disease are described per year.8 18 19 26 28 35 36 Juvenile and late onset Blount disease is even more uncommon.15 28 With 139 Blount patients in 8 years, this study presents the highest prevalence of Blount disease patients in the literature up to date.
Genetic contribution and familial influence were suggested in earlier studies but has not yet been extensively studied.5 9 10 19 27 Table 3 recapitulates the literature on familial occurrence in early and adolescent Blount disease. The study data show a higher prevalence of positive family history for varus legs in all groups, compared with the literature.
Articles on familial in occurrence Blount disease
Research in Ghana can be challenging and therefore some limitations of this study need consideration. First, data are based on self-reported family history. But, to prevent different sides of the family accusing each other, we always needed confirmation from both sides of the family. The exclusion of rickets and polio was also based on self-reported data. Second, due to the absence of electronic patient files and digital X-rays before February 2015, we had to exclude 22 possible Blount disease patients because of missing files and, or X-rays. Finally, low resources and expensive journeys to a hospital could prevent patients from seeking medical consultation. In addition, having varus legs has some aesthetic value in Ghana. This could explain why nine out of the 12 cases with severe varus legs in the family history never visited the hospital for medical consultation.
In conclusion, this study describes a high population of Blount disease. Because of the high numbers of positive family history for varus legs, especially in the early onset group and the centralisation of patients in the Akan region, a familial predisposition for varus legs is probable. Our data are consistent with the hypothesis that Blount disease is primarily the result of the proximal tibial epiphysis responding to physical phenomena, in which varus alignment has a greater mechanical effect compared with weight. Therefore, the high prevalence of familial inheritance for varus legs and its mechanical effect on the medial physis of the proximal tibia might explain the large population of Blount patients found in this study, in a predominantly non-overweight population.
The first analysis of a routine WES-based diagnostic approach for variant filtering and selection while interrogating the entire exome and while systematically comparing the exome sequencing data between patients from different families did not show a genetic predisposition. Based on the family tree’s and the WES outcome, a Mendelian inheritance was not found. Nonetheless, this study shows optimistic results for deeper interrogation of the genome using less stringently filtered raw sequencing data in a research setting. Also, more research on familial occurrence in the juvenile and late onset Blount patients is needed, because these studied groups are rather small compared with the early onset group.
Data availability statement
Data are available upon reasonable request at the corresponding author.
Acknowledgments
We are very grateful for the help Hussein Botchway provided us to get approval from the Medical Ethical Committee of Kwame Nkrumah University Kumasi (Ghana). Furthermore, we would like to thank the whole staff St. John of God hospital in Duayaw Nkwanta (Ghana) for helping out in every possible way to make this research possible.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors NJ conceptualised the study, performed the investigation, wrote, reviewed and edited the manuscript. FH conceptualised and supervised the study, wrote, reviewed and edited the manuscript. FB collected data. PM acquired the resources, supervised the study. AS analysed data. HMS conceptualised, acquired the resources and supervised the study.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Map disclaimer The depiction of boundaries on the map(s) in this article does not imply the expression of any opinion whatsoever on the part of BMJ (or any member of its group) concerning the legal status of any country, territory, jurisdiction or area or of its authorities. The map(s) are provided without any warranty of any kind, either express or implied.
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.