Article Text

Abstract
Introduction One in three Danish children under 3 years of age experience asthma-like symptoms, and one-third will later be diagnosed with asthma. Oral prednisolone is used in various formulations to treat acute asthma. However, the potential differences in bioequivalence between these formulations have never been examined in children despite interchangeable use in clinical practice.
Methods and analysis An open-label, randomised, two-treatment cross-over trial investigating the bioequivalence of different prednisolone formulations in children with airway disease.
The included patients (6 months–11 years of age) are admitted to the Department of Paediatric and Adolescent Medicine Nordsjællands University Hospital, Hillerød, with asthma or asthma-like symptoms.
The primary objective is to assess the bioequivalence between different prednisolone formulations herein area under the concentration time curve, Cmax and Tmax using saliva samples. The secondary objectives are to evaluate tolerability (five-point face scale), adverse events and severity of the disease. If the patient has an intravenous access for other purposes, the saliva samples will be validated with plasma samples.
A total of 66 evaluable patients are needed according to European Medicines Agency Guideline on bioequivalence.
Ethics and dissemination Traditional pharmacokinetic trials are burdensome due to the extent of blood samples necessary to capture the time-dependant drug profile. Saliva sampling is far more acceptable for paediatric patients. In addition, this trial adheres to standard dosing strategies. No additional venepunctures are performed, and no additional prednisolone doses are administered.
Guidelines for paediatric bioequivalence trials are warranted.
Trial registration number The Danish Medicines Agency EudraCT: 2017-003590-33, The Ethics Committee case no: H-17027252, and the Danish Data Protection Agency: BFH-2017–103, I-Suite no.: 05935.
- respiratory
- allergy
- pharmacology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic?
Various prednisolone formulations are frequently used to treat asthma-like symptoms in children; the potential differences in bioequivalence between these formulations have never been examined.
Pharmacokinetic studies have traditionally been burdensome for the paediatric population due to numerous blood samples.
It is important to study the acceptability of paediatric formulations, since manipulating medicine or introducing different formulations may diminish or enhance tolerability.
What this study hopes to add?
Paediatric bioequivalence data on prednisolone oral solution, crushed tablets and orodispersible tablets compared with whole tablets.
Prednisolone concentration data are collected using saliva samples, validated with plasma samples if the patient has an intravenous access for clinical purposes.
Acceptability data on different prednisolone formulations using a modified Wong-Baker face scale.
Introduction
One in three Danish children under 3 years of age experience asthma-like symptoms, and one-third will later be diagnosed with asthma.1 Therapeutic management of acute asthma-like symptoms in infants and children has been repeatedly discussed.2 Oral glucocorticoids are frequently added to inhaled beta-2-agonist if the patient does not respond to the initial treatment. However, no therapeutic interval exists for glucocorticoids, and no clinical biomarker has been deveopled to asses efficacy.3
Furthermore, in Denmark, only intravenous formulations and prednisolone or hydrocortisone tablets are licenced for patients <18 years of age.4 Infants and children who are unable or unwilling to swallow a tablet or where dosing is based on body weight are urged to use either an extemporaneous oral liquid formulation, crushed or split tablets. Splitting tablets may lead to dose inaccuracy, and in one study, crushing hydrocortisone tablets lead to therapeutic failure.5 Extemporaneous formulations are not subject to randomised controlled trials as licenced formulations and bioequivalence (BE) are not determined between different formulations with the same active ingredience.5 6 Moreover, the paediatric population is a heterogeneous group of patients ranging from premature neonates to adolescents. The wide physical and developmental differences influence both pharmacokinetic (PK) and pharmacodynamic parameters. These issues are generally poorly studied.7
BE studies compare both the rate (the time to Cmax=Tmax) and extent of absorption (area under the concentration time curve (AUC) of various drug formulations. Regulatory guidelines on BE state that two treatments are not different from one another if the 90% CI of the ratio of a log-transformed exposure measure (AUC and/or Cmax) falls within the range of 80%–125%.8
To date, one BE study with prednisolone have been performed in healthy adults.9 Francisco et al9 examined AUC, Tmax and Cmax of five commercially oral prednisone tablets compared with one reference solution in 18 healthy adults.9 No statistically significant differences were found between the tablets. Non-significant variations were seen between the solution and tablets except for Tmax, which was 0.53 and 1.2–1.7 hours, respectively.9 A bioavailability study from Georgitis et al10 compared liquid prednisone, prednisone tablet and intravenous prednisolone10 and found that the AUC for liquid and tablet were significantly different.10 No studies have been performed in a target patient population, taking into consideration factors such as comorbidities, comedication and physiological factors including first pass metabolism, gastric pH and bacterial flora, which are highly relevant in infants and children.
However, traditional PK trials are burdensome due to the extent of blood samples, necessary to capture the time-dependant drug profile. New non-invasive methods such as saliva sampling are far more acceptable for paediatric patients. In a recent adult population PK study, free prednisolone concentrations correlated well with salivary concentrations.11 The same study set-up were later applied in a population PK model in paediatric patients (n=104) with nephrotic syndrome.12 None of the studies included more than one formulation.
The importance to study the acceptability of paediatric formulations has recently been endorsed since manipulating medicine or introducing different formulations may diminish or enhance tolerability.13 Acceptability of medicines influence adherence to therapeutic regimens and outcomes. No validated method to access acceptability of paediatric medicine exists, and many different scales have been used most commonly hedonic face scales or visual analogue scale (VAS).13 Lucas-Bouwman et al14 investigated the acceptability of prednisolone oral solution and crushed tablets using VAS taste scale. The oral solution obtained the best score and no vomiting was observed in the oral solution treatment group compared with 23% in the other group. Aljebab et al15 showed that dexamethasone elixir was more palatable than prednisolone tablets, soluble tablets and syrup using af five-point hedonic face scale in children below 12 years of age in the UK and Saudi Arabia.15 Prednisolone syrup and soluble tablets caused less vomiting than tablets.15
Objectives
We aim to investigate the bioeqvalence of different prednisolone formulations in three different age-groups and secondary to investigate the tolerability, adverse events, severity of the disease and validation of saliva samples, table 1.
Primary and secondary objectives and outcomes in the POP child trial
Methods and analysis
A single-centre, randomised, open-label two-treatment cross-over design.
Children with asthma or asthma-like symptoms admitted to the Department of Paediatric and Adolescent Medicine, Nordsjællands University Hospital, Hillerød, are eligible for inclusion.
Population
Eligibility criteria are described in table 2. Patients are stratified by age in accordance with European Medicines Agency (EMA) guidelines into three groups, that is, 6–23 months, 2–5 years and 6–11 years.16 Two different formulations of prednisolone as 1 mg/kg/day will be administered to each participant for two consecutive days (figure 1). A control group of school children age 6–11 years will receive whole tablets. In total, four different formulations will be investigated (see figure 1). All trial medicine is labelled, packed and distributed by the Regional Pharmacy in accordance with GMP Annex 13,17 except the high-strength oral prednisolone solution 20 mg/mL, which is ‘ward medicine’ and does not require special labelling. A separate accountancy is kept for each formulation of prednisolone.
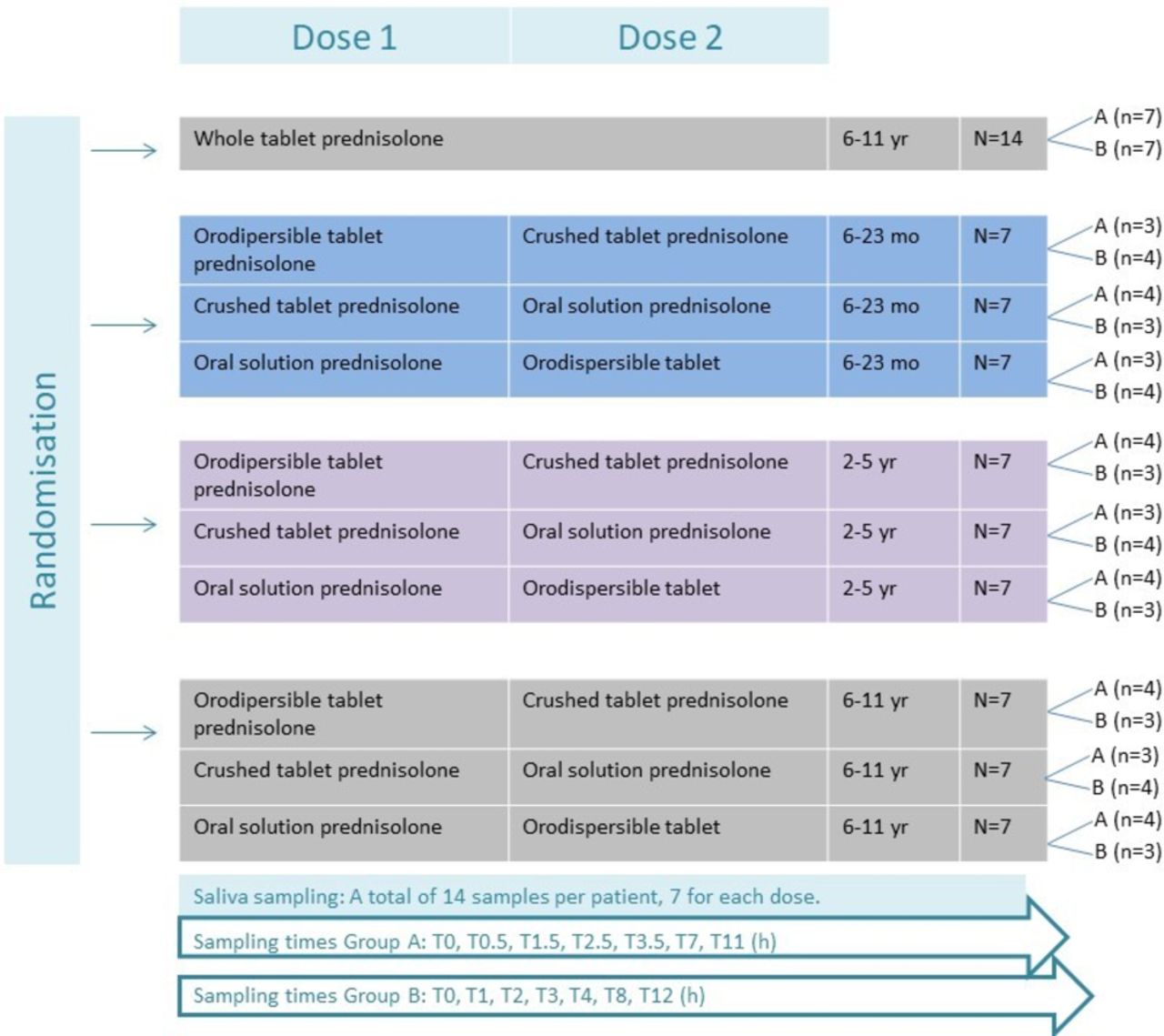
In total, four investigational medical products (IMPs) are used: ‘Prednisolon DAK’ whole tablets, ‘Prednisolon DAK’ crushed tablets (5 or 25 mg), extemporaneous prednisolone oral solution and SOLUPRED orodispersible tablets (5 or 20 mg imported from France). The oral solution exists in two strengths, that is, 5 mg/mL (for children below 10 kg) and 20 mg/mL (for children above 10 kg).
Inclusion and exclusion criteria for the POP child trial
Patient involvement
Patients were not directly involved in the design of this study.
Saliva samples
The primary and secondary outcomes are assessed at predefined time points shown in table 3 and figure 1. Patients are randomised to one of two sampling groups, A and B, figure 1. Sampling times are calculated on the basis of a previous study.12 Each child will follow the same time schedule for both doses. The samples will be collected using the SalivaBio Children’s Swab and the SalivaBio Oral Swab (exclusively from Salimetrics, State College, Pennsylvania, USA), synthetic swabs specifically designed to improve volume collection and increase participant compliance. Saliva will be removed from the swabs by centrifuge (15 min at 2500 rpm) and stored in cryovials at −20 ᵒC for maximum 2 weeks and hereafter at −80°C until analysis. Concentrations of prednisolone and its inactive metabolite prednisone will be determined by liquid chromatography with mass spectrometric detection. The assay is validated according to the EMA guideline ‘Guideline on bioanalytical method validation’ (EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2**Committee for Medicinal Products for Human Use). Analyses are performed in accordance with ISO 15189. Concentrations will be determined in 10 µL saliva or plasma. In the latter case, both total and unbound concentrations will be determined. The lower limit of quantification is 2 ng/mL for both compounds. Saliva samples are non-invasive samples and are considered a painless low-risk procedure.16 Saliva sampling of prednisolone and subsequent analysis has recently been performed in children with median 4.6 years of age using Salivette equipment.12 As this trial includes children from 6 months of age, we chose the Salimetrics equipment. If any child has a cubital intravenous access at inclusion, additional blood samples will be taken time equivalent to the saliva samples. In order to assess plasma prednisolone for validation of the saliva samples. No additional venepunctures will be made.
The table illustrates the different trial activities.
Sample size
The number of subjects required for a BE study depends on the expected deviation between formulations.18 Because of limited data on prednisolone PKs and BE of different formulations, sample size calculation is not feasible. In accordance with the EMA guideline on the investigation of BE,8 the number of subjects to be included in a study should preferably be based on sample size calculations and not be less than 12. To account for drop-outs, 14 patients were allocated to each formulation in the three different age groups and the control group (a total of 77 patients) (figure 1). The optimal number of participants is 66 evaluable patients.
Randomisation
The participants are randomised in the REDCap software (www.project-redcap.org). The REDCap software is mandatory to use in investigator-initiated research in the Capital Region in Denmark. The randomisation list is uploaded in REDCap and is blinded to everyone except a third person, who generates the list and has no other role in the trial. The participants are age stratified and will be block randomised to ensure an equal amount in each subgroup (figure 1). The child will receive a REDCap generated ID number including a combination of letters describing the formulations to be given. Administration of prednisolone is done open-label by the project staff or paediatric nurses.
Statistical methods
The AUC(0 t) and Cmax parameters will be calculated using single subject compartmental modelling. Actual time of sampling will be used in the estimation of the PK parameters. The assessment of BE will be based on 90% CIs for the ratio of the population geometric means (test/reference), with the null hypothesis of BE at the 5% significance level (acceptance interval of 80.00%–125.00%)8 using ANOVA or t-test. The comparison between the different age groups will be done in a stepwise manner, initially comparing the various subpopulation, for example, the 6–23 months old. If these data are comparable, we will pool the data and compare with the older age groups. The oldest age group will only then serve as control group across age population. If the data do not show BE, the bioavailability data will be reported separately for each age group.
The person responsible for the statistical analysis of saliva samples will be blinded to the formulations given. Due to the limited wash-out period, a test for carry-over will be addressed by examination of the pretreatment plasma concentrations in period 2.
All patients who fulfils the eligibility criteria and have had at least one saliva sample taken will be analysed for demographic and baseline data. In the PK modelling, all saliva samples will be included. If any predose concentration is more than 5% of Cmax, this will be reported and handled appropriately in the final analysis.
Ethics and dissemination
Before prednisolone treatment is initiated, a patient information sheet is given to eligible patients and the trial is explained to their parents. Parents will have a minimum of 15 min to consider the consent, because treatment should not be unnecessarily delayed. Informed consent will be granted by parents or legal guardian, as all children are below 15 years of age. If one of the parents is not present, a written authority can be used. The consent process will be conducted by an investigator with training and experience with minors. The collected material is considered a research biobank.
Safety
Despite a large therapeutic index, it is important that all glucocorticoids are administered in the lowest effective dose as possible, since a considerable number of dose-dependent adverse drugs reactions (ADRs) are well described.4 A recent review found that the most frequent ADRs associated with short course oral corticosteroids (<14 days) in children were vomiting, changes in behavoiur and sleep disturbances with incidence rates of 5.4%, 4.7% and 4.3%, respectively. Increased susceptability to infection were one of the serious ADRs ocurring in up to 1% of the children.19
Trial limitations
Alterations have been made in this trial compared with a classic BE trial to adapt to the relevant population. There are no existing guidelines for paediatric BE trials. A guideline is warranted since paediatric BE trials must be pragmatic in order to adapt to the paediatric population and to be conducted ethically.
Optimally, a randomised, two-period/three-period, two-sequence/three-sequence single-dose cross-over design would be used for healthy adults. This trial is a single-dose study performed during admission, limiting the possible number of doses/formulations per patient, with a theoretical washout period of 24 hours, as prednisolone is dosed once daily. The half-life of prednisolone is 2.5±0.5 hours,20 21 and 24 hours should be enough time to ensure drug concentrations below quantification. However, as a precaution, predose samples are collected if prednisolone has been administered the day before inclusion. The saliva samples will be measured in 11 or 12 hours, respectively, which will be within five half-lives for many patients. Late night samples will be more difficult to collect if the participants are asleep and some might not be collected due to patient and parent preferences.
The reference product in this trial is whole tablets, since intravenous glucocorticoids are mainly used for severe asthma. The effect of fasting and fed are not within the scope of this trial since no effect on bioavailability was found in prednisolone whole tablets.4 To avoid prednisolone leftovers in the mouth, all children will be given water after administration of trial medicine. There can be differences in which the hedonic face scale is presented to the child, and this can be of some limitation (figure 2). Only 2–5 persons will include participants in this trial, minimising the variability.

{kind=link}
{kind=link}
Modified Wong-Baker face scale to access tolerability of trial medicine. Self-reported tolerability of the medicine is collected from children older than 6 years of age. The options A–E is read aloud neutrally to the child. For children below 6 years of age, a parent is asked to evaluate the tolerability. Vomiting is registered separately but is included in the face scale as F.
We expect to have some dropouts due to: (1) non-compliance with the formulations and (2) due to earlier than expected discharge. In the control group with whole tablets data suggest that about 91% of 6-year-old children can swallow a 7 mm tablet.13 The whole tablets are 8 mm (5 mg) and 10 mm (25 mg) but with break lines. We chose to include two strengths of prednisolone oral solution, to make sure the volumes were measurable and acceptable.13 Dropouts and inclusion only during daytime could give some recruitment bias; however, as this is primarily a PK trial, we do not expect this to influence the results. This trial is conducted at one site which can limit the generalisability but will enhance the consistency.
Trial registration
This trial is an investigator-initiated trial from the Department of Clinical Pharmacology, Bispebjerg Hospital and the Department of Paediatric and Adolescent Medicine, Nordsjællands Hospital, Hillerød. The trial is approved by the Danish Medicines Agency EudraCT: 2017-003590-33, the Ethics Committee case no: H-17027252 and the Danish Data Protection Agency: BFH-2017–103, I-Suite no.: 05935. The trial is monitored by the GCP-unit Copenhagen University.
References
Footnotes
Contributors HH, SSH-K and IMJ were involved in the conception and design of the study. HH, SSH-K and IMJ wrote the protocol and applied for all permissions and funding. All authors were involved in preparing the study sites, acquisition of data and analysis, in writing the protocol article and in its revision prior to submission.
Funding The salary to the PhD student is covered by Department of Clinical Pharmacology and the Danish Regions. Costs for trial medicine, project staff and analysis of saliva samples are covered by the A.P. Møller Foundation, Captain Lieutenant Harald Jensen and Wife Foundation, The Danish Medical Research Grant and Aase and Ejnar Danielsens Foundation.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The article reports a protocol for an ongoing trial. Therefore, no trial data are available yet. All data relevant to the study are included in the article or can be proqured from the authors upon requst.